Downloaded 1,178 times

















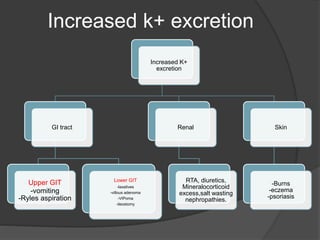





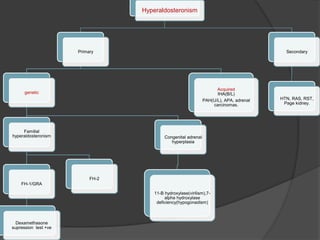








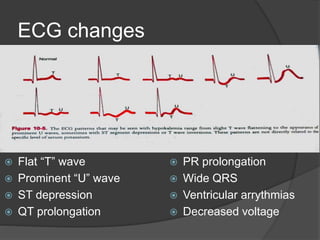


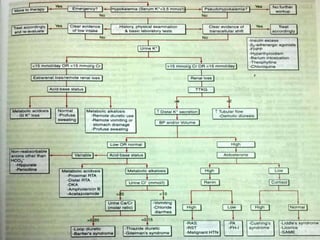










This document discusses hypokalemia, defined as a serum potassium level below 3.6 mEq/L. It notes that potassium is critical for nerve impulse transmission and muscle function. The major causes of hypokalemia include decreased intake, intracellular shifts, non-renal losses like vomiting/diarrhea, and renal losses due to drugs like diuretics. Clinical features range from muscle weakness to cardiac arrhythmias. Diagnosis involves evaluating electrolytes, ECG changes, urine studies, and considering underlying causes. Treatment focuses on replacing potassium losses and correcting the underlying etiology.



































































![potasium disorNNNNNNNNNNNders [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/potasiumdisordersautosaved-240622073502-4241000d-thumbnail.jpg?width=640&height=640&fit=bounds)
















![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


