
Downloaded 15 times







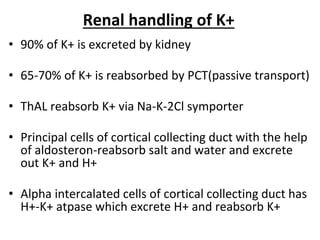
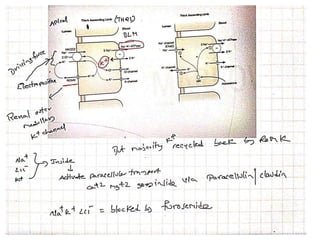


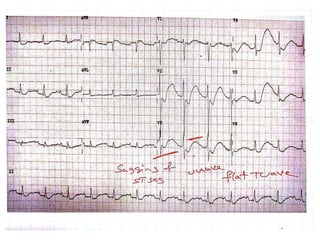
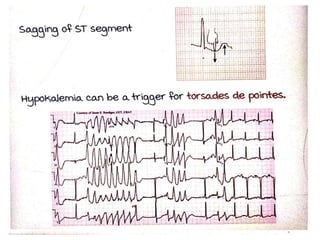

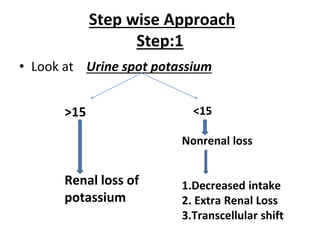

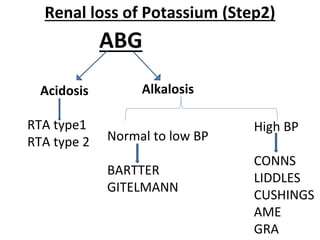



















This document discusses the physiology and pathophysiology of hypokalemia. It covers the normal distribution and regulation of potassium in the body, as well as causes of hypokalemia including decreased intake, extrarenal losses, and renal losses. Types of renal tubular acidosis that can cause hypokalemia are described. The approach to hypokalemia involves considering underlying causes like Bartter syndrome, Gitelman syndrome, Liddle's syndrome, and hypomagnesemia. Treatment principles for correcting hypokalemia are provided.















































































