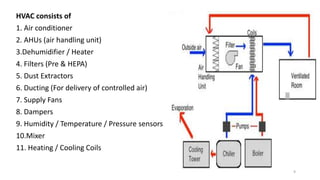
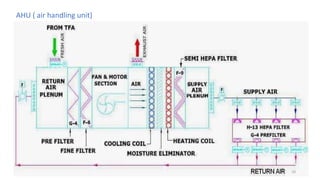
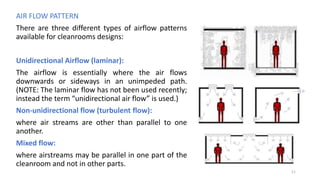
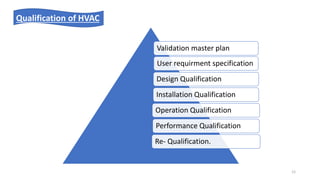
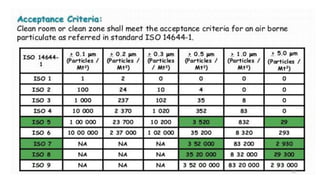
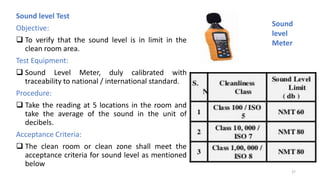
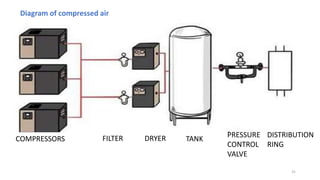
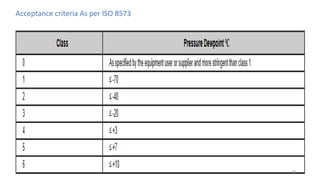
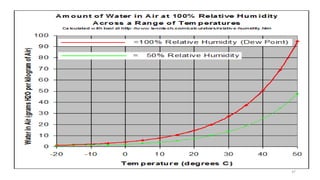
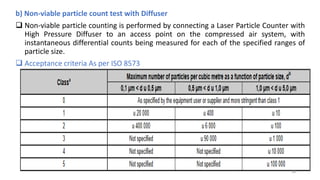
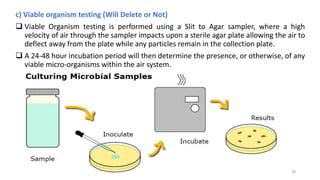
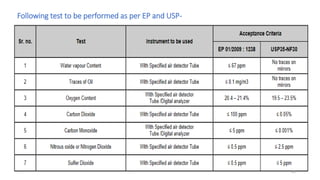
The document provides information about validating the HVAC system in a pharmaceutical facility. It discusses the importance of HVAC systems in maintaining suitable temperature, airflow and preventing cross-contamination. It describes the key components of an HVAC system and various validation parameters to test, including air flow measurement, filter integrity testing, particulate counting and temperature/humidity levels. The validation process involves design qualification, installation qualification, operational qualification and performance qualification of the HVAC system to ensure it meets predetermined specifications.