Download as PDF, PPTX


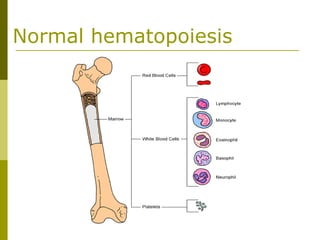
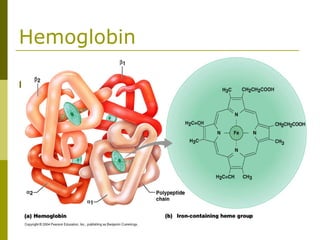










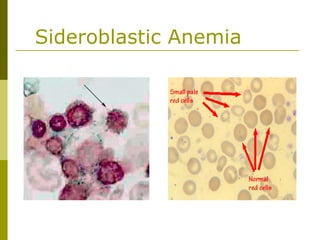

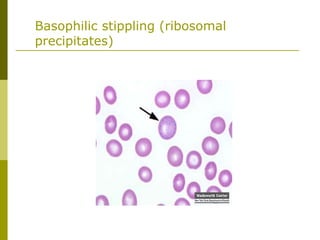
















































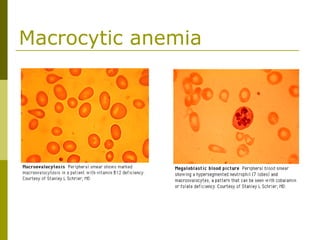

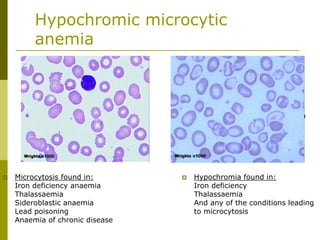

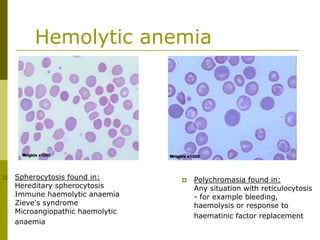
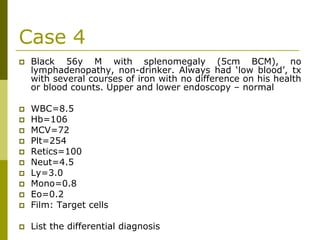






This document provides an overview of hematology and approaches to common blood disorders. It defines anemia and describes how to classify it based on mean corpuscular volume. Common causes of microcytic, macrocytic, and normocytic anemia are outlined. Diagnostic criteria and treatments for iron deficiency anemia, anemia of chronic disease, and other specific anemias are summarized. The document also reviews approaches to thrombocytopenia, neutropenia, pancytopenia, and transfusion reactions.


























































































