Download to read offline






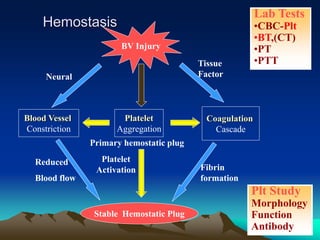




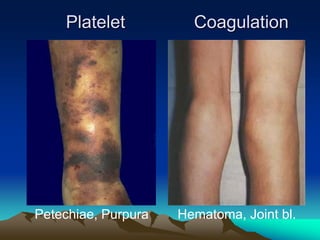




















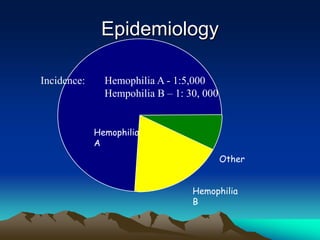



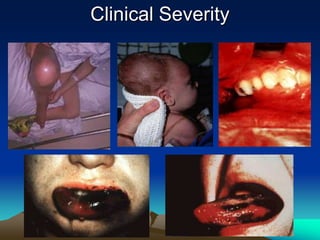
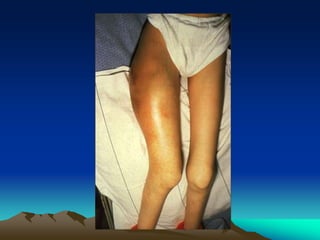
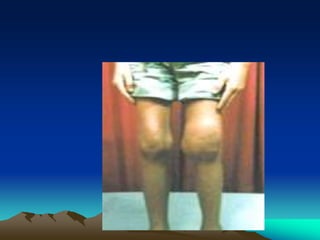






Hemorrhagic disorders involve problems with hemostasis, the process of stopping bleeding. There are bleeding disorders which can be caused by platelet defects or vascular defects. Hemorrhagic disorders involve deficiencies in coagulation factors. Common hemorrhagic disorders include hemophilia A which is a factor VIII deficiency and hemophilia B which is a factor IX deficiency. Both hemophilia A and B are inherited disorders that primarily affect males. Treatment involves replacement of the deficient coagulation factor through products such as fresh frozen plasma or concentrated factor products.



































































