Downloaded 142 times





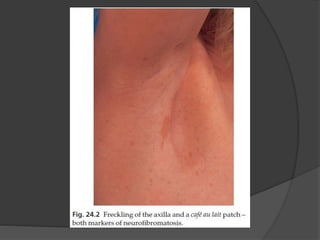





















Genodermatosis refers to inherited skin conditions caused by genetic mutations. This document summarizes information on three types: neurofibromatosis type 1 (NF1), tuberous sclerosis, and xeroderma pigmentosum. NF1 is caused by mutations in the NF1 gene and is characterized by café au lait spots, freckling, neurofibromas, and Lisch nodules. Tuberous sclerosis results from mutations in TSC1 or TSC2 and presents with ash leaf macules, angiofibromas, and connective tissue nevi. Xeroderma pigmentosum involves defective DNA repair causing photosensitivity and early development of skin cancers upon






































































































