Download as PDF, PPTX























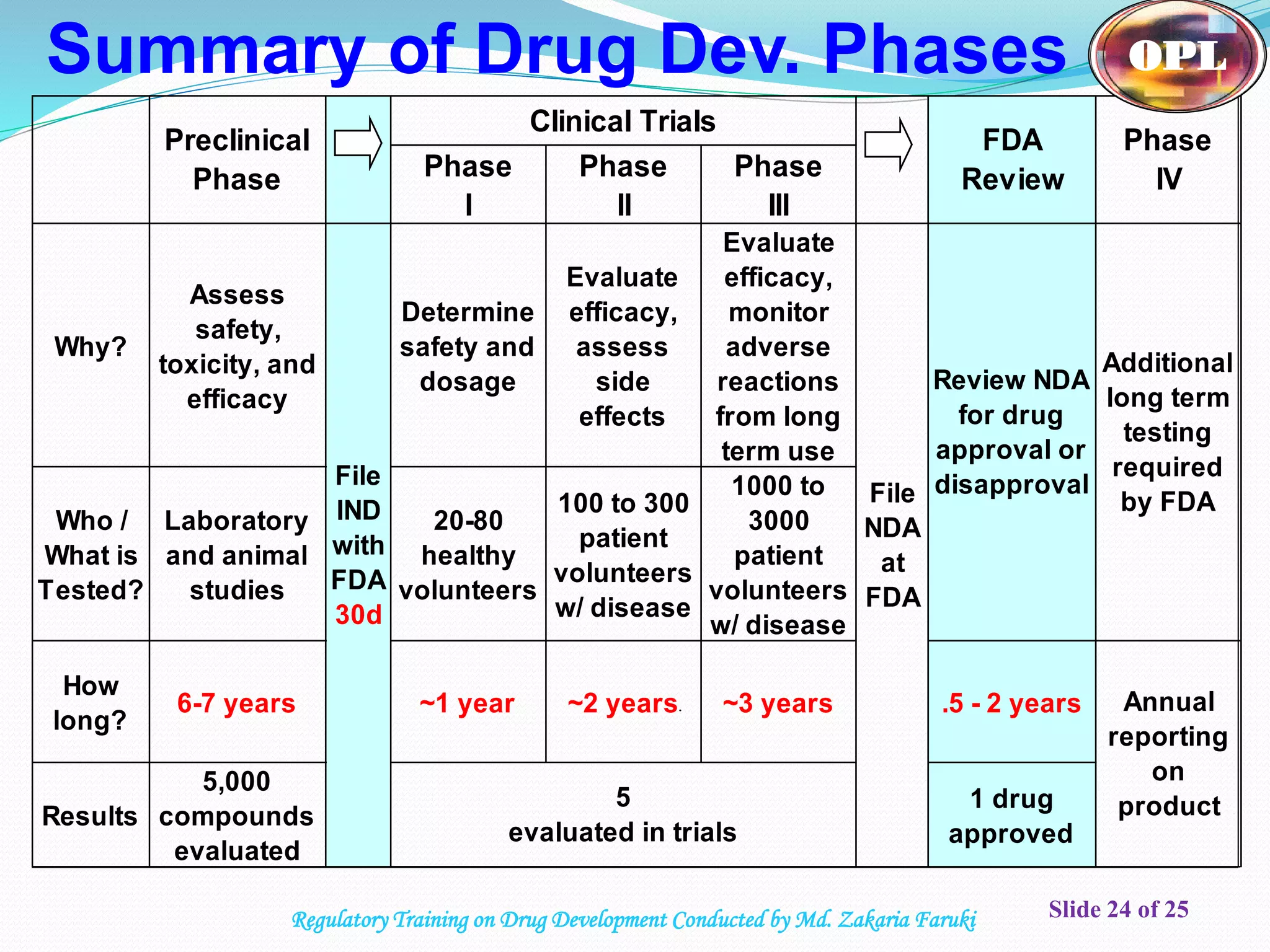


The document discusses the process of drug development from discovery through clinical trials and regulatory approval. It begins with an overview of the iterative process involving biology, animal testing, and medicinal chemistry. It then outlines the typical 5 steps in drug development according to the FDA: discovery/screening, pre-clinical research including in vitro and in vivo testing, clinical research consisting of Phase I-III trials, FDA review, and post-marketing monitoring. The document provides details on each stage of development including pre-clinical and clinical research requirements and processes.











































![Preclinical_Phase_of_Drug_Development_Lucas[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/preclinicalphaseofdrugdevelopmentlucas1-251107071109-7c5cab19-thumbnail.jpg?width=640&height=640&fit=bounds)











































