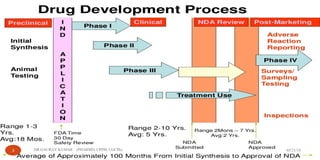
The document outlines the 5 step drug development process: 1) discovery and development, 2) preclinical research, 3) clinical research consisting of 4 phases, 4) FDA drug review, and 5) post-market safety monitoring. It provides details on each step, including discovery methods, preclinical toxicity testing in animals, clinical trial design, and FDA requirements and oversight at each phase of development. The overall process takes an average of 10-15 years from discovery to approval due to the extensive testing required to demonstrate safety and efficacy.













































![INVESTIGATIONAL NEW DRUG [IND] APPLICATION SUBMISSION (1).pdf](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugindapplicationsubmission1-221128141339-adad00bd-thumbnail.jpg?width=640&height=640&fit=bounds)
















































![Clinical trial public access registries [recovered]](https://cdn.slidesharecdn.com/ss_thumbnails/clinicaltrialpublicaccessregistriesrecovered-190521115916-thumbnail.jpg?width=640&height=640&fit=bounds)



















