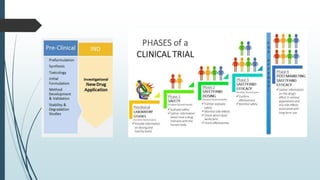
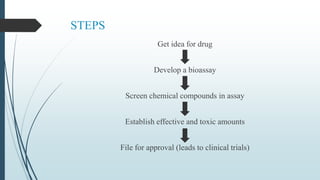
The document discusses pre-clinical trials, which are essential studies conducted on animals to test the safety and efficacy of new drugs before human trials. It explains different types of pre-clinical trials, including in vitro and in vivo methods, and outlines the stages, goals, and considerations involved in these studies. The ultimate aim is to ensure that promising treatments are safe for human testing by collecting vital safety data and identifying initial effective dosages.






















































![Preclinical_Phase_of_Drug_Development_Lucas[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/preclinicalphaseofdrugdevelopmentlucas1-251107071109-7c5cab19-thumbnail.jpg?width=640&height=640&fit=bounds)
























