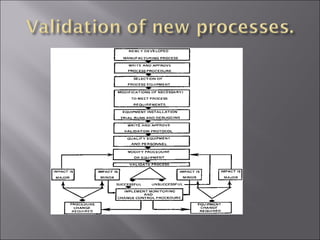
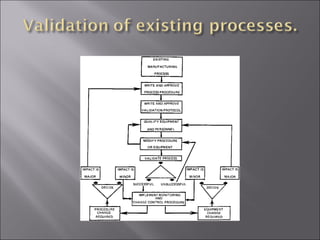
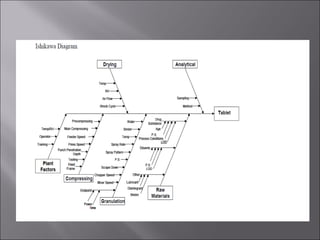
The document discusses key aspects of validating solid dosage forms such as tablets. It emphasizes that quality must be built into the product from the beginning, starting with validating the characteristics of the active pharmaceutical ingredients and excipients used. Analytical methods, manufacturing equipment, and the entire production process must also be validated to ensure reproducible quality batches. The validation program involves defining critical material attributes, establishing control parameters, and testing batches to set specification limits to maintain process control.