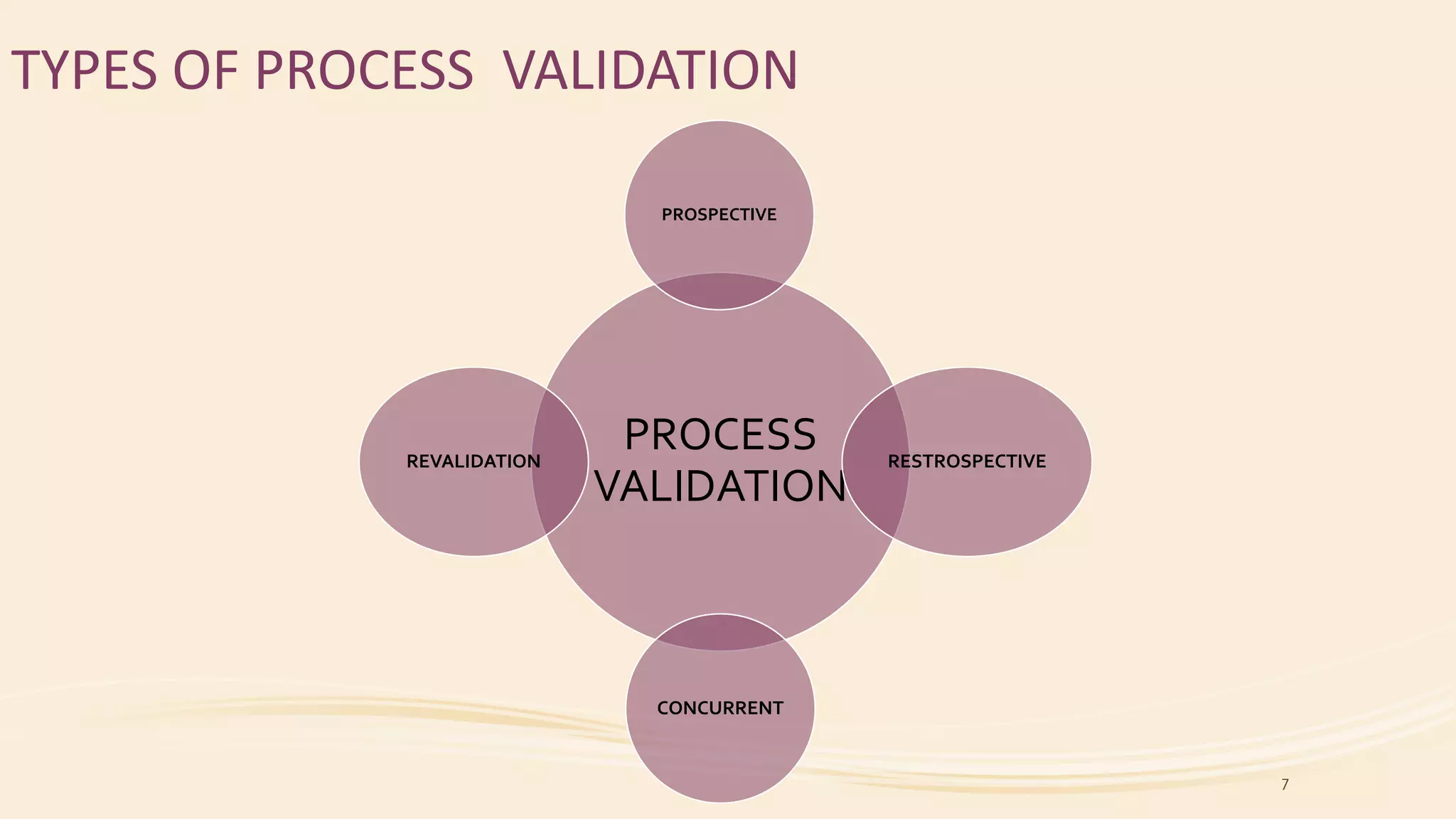
This document discusses types of process validation for pharmaceutical manufacturing. It describes prospective, retrospective, and concurrent validation. Prospective validation involves validating a new process before use by following a validation protocol. Retrospective validation analyzes historical data from established stable processes. Concurrent validation monitors critical parameters during production. The document also discusses validation of tablet and capsule manufacturing processes. Key parameters include composition, mixing/blending, granulation, drying, milling, lubrication, compression/filling, and testing. Process validation ensures consistent quality and safety of dosage forms.