

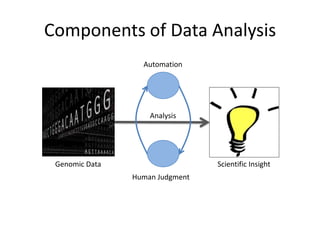


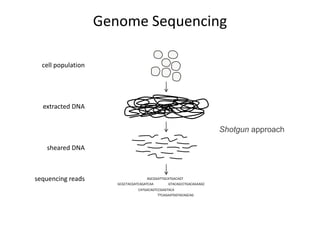
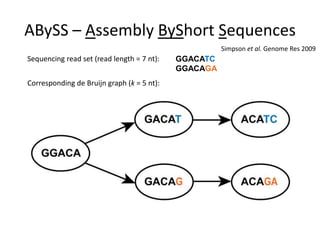
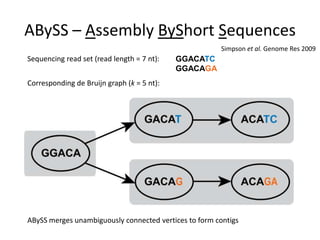
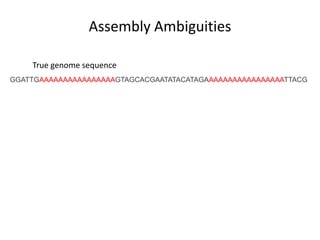
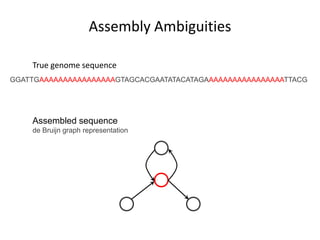
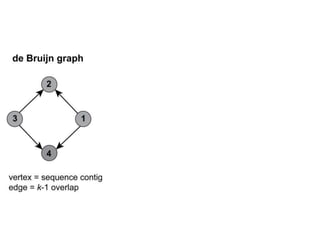
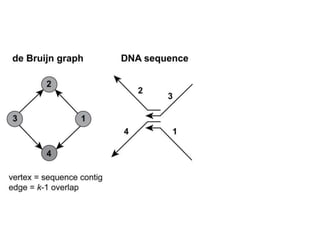
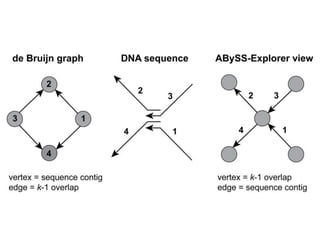

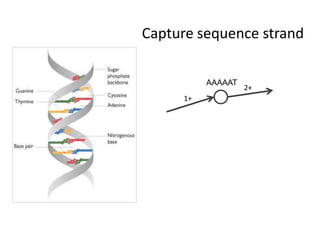
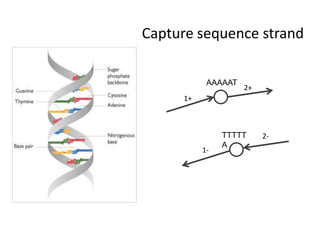
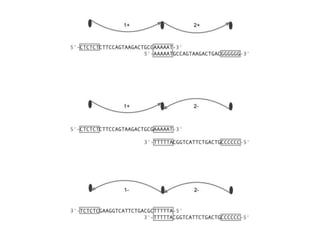
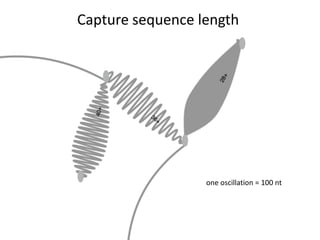
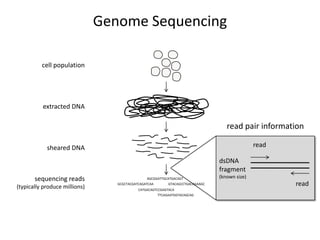
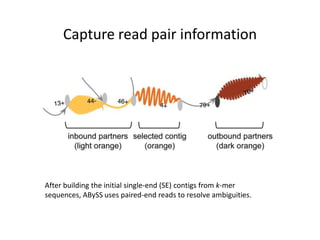
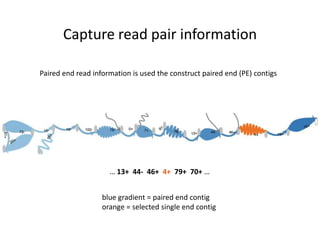




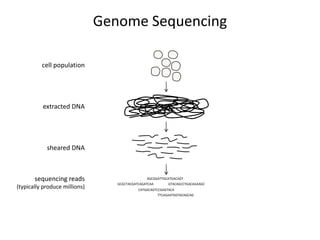
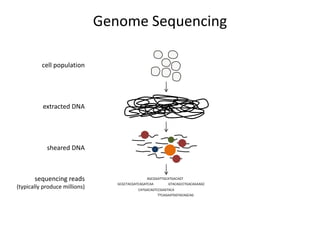
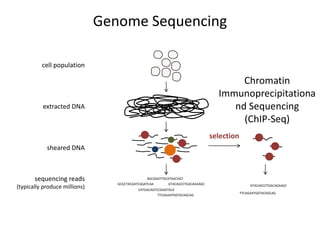
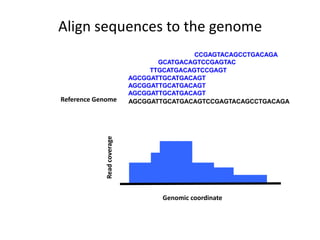
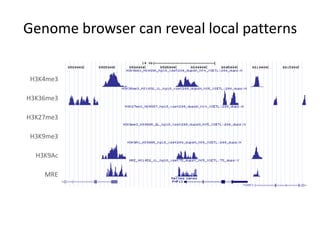
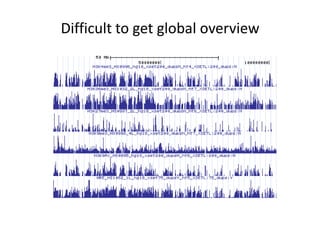


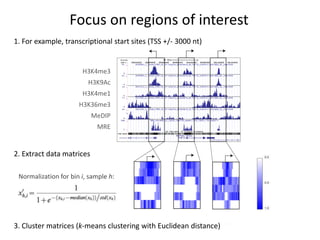
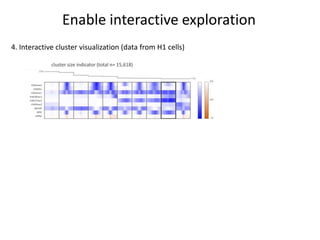
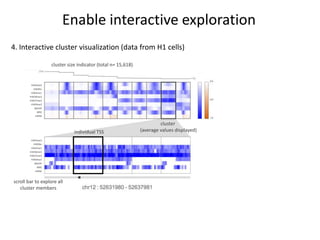
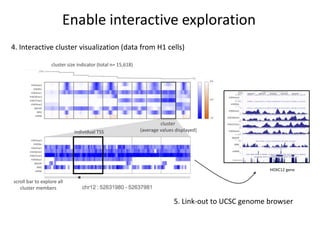



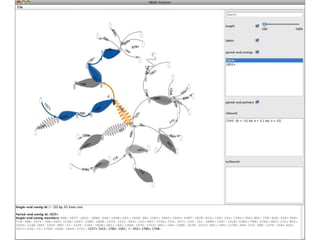
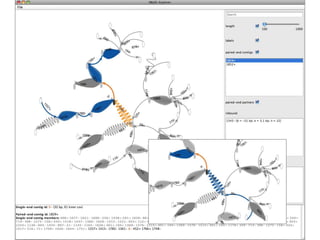


The document discusses the integration of computation and visualization in genomics, particularly focusing on the use of the software 'abyss-explorer' for genome assembly visualization. It highlights the processes for genome sequencing, resolving ambiguities through paired-end reads, and the effectiveness of clustering for revealing patterns in genomic data. Future work aims to enhance the tool's functionality, including search capabilities and richer analysis features.









































































