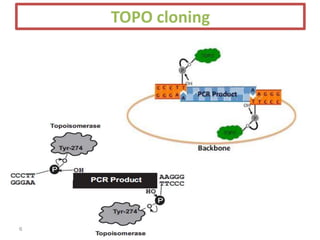
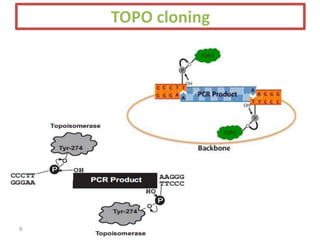
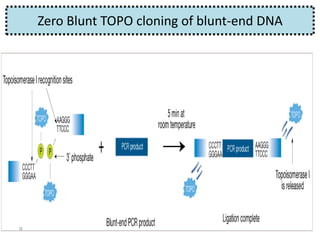
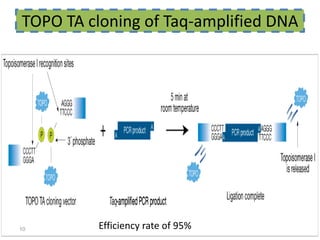
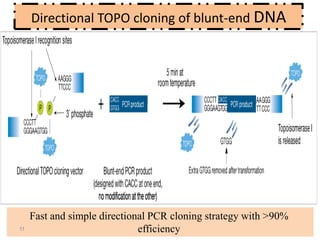
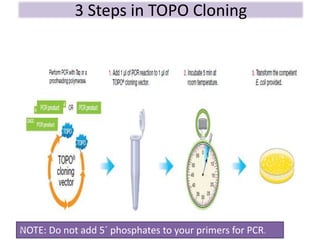
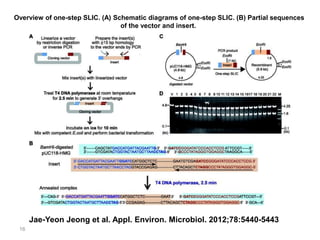
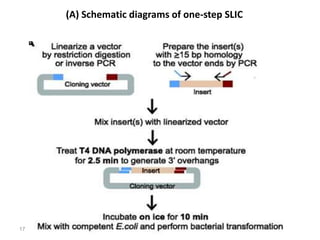
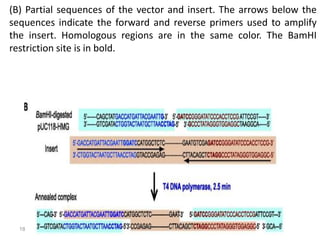
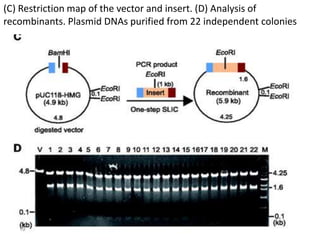
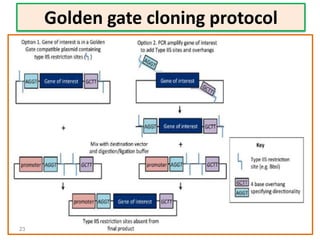
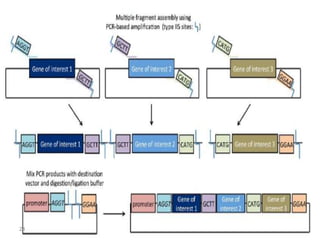
The document discusses various DNA cloning techniques, including topo cloning, sequence and ligation-independent cloning (SLIC), and golden gate cloning, along with their characteristics and procedures. It highlights the importance of vectors in genetic engineering and provides insights into the efficiency and application of these techniques. Each method is evaluated for its advantages and disadvantages, along with references for further reading.