Downloaded 414 times







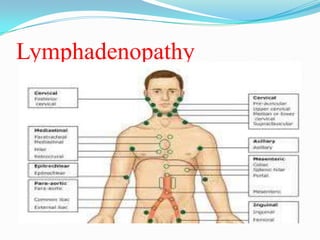
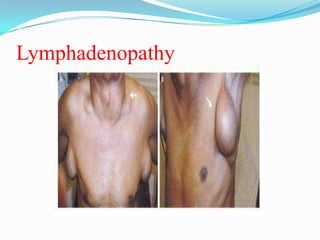

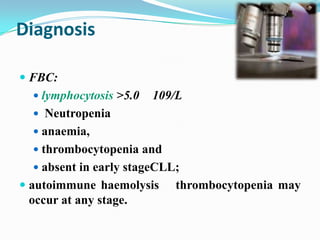
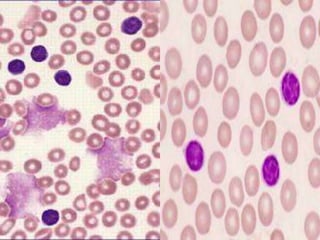

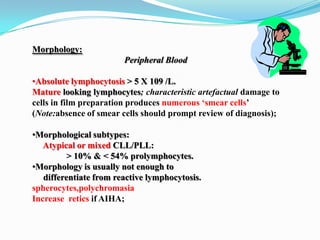
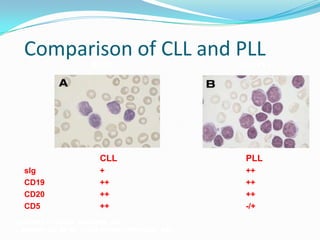



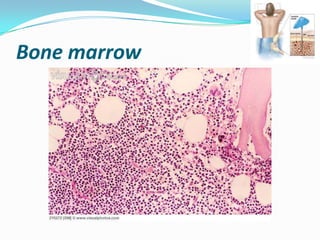
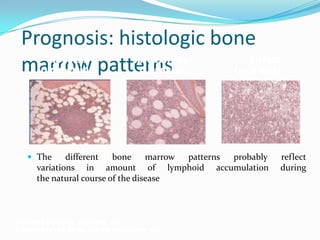










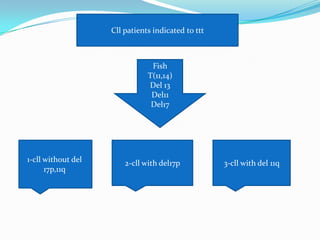
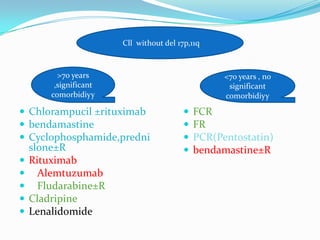



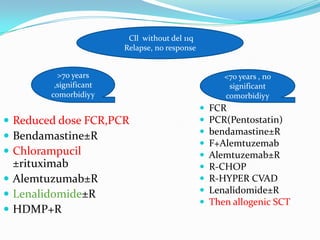




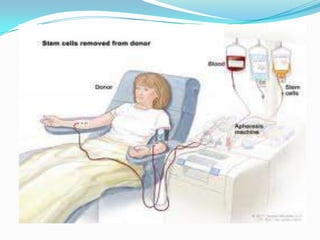





This document provides information on chronic lymphocytic leukemia (CLL), including its definition, diagnosis, incidence, aetiology, clinical features, diagnostic tests, staging systems, prognostic factors, and treatment approaches. Some key points include: - CLL is defined as the progressive accumulation of long-lived, functionally incompetent B lymphocytes. - Diagnosis involves evaluating lymphocyte counts, immunophenotyping, bone marrow biopsy, and cytogenetic/molecular testing. - Incidence is highest in Western adults over age 65 and is more common in men. - Prognostic factors include genetic abnormalities, IgV gene mutation status, and response to initial therapy. - Treatment depends on















































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)






