Download as PDF, PPTX





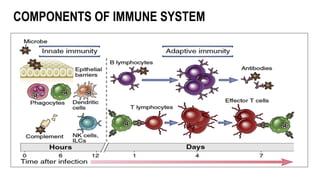
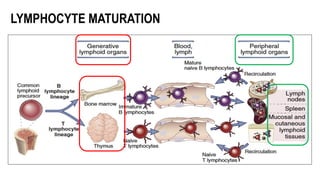

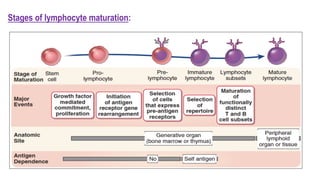
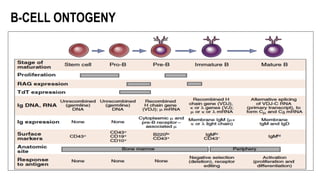
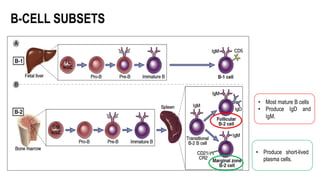
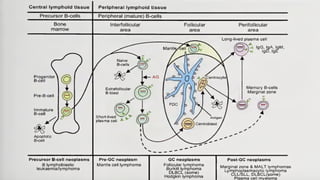
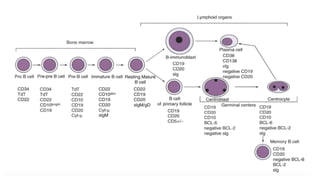
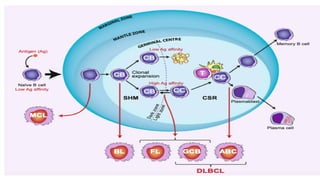
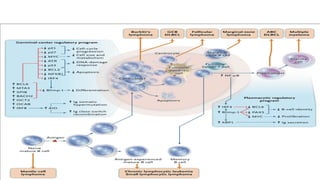




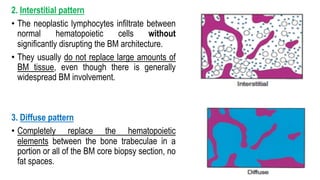
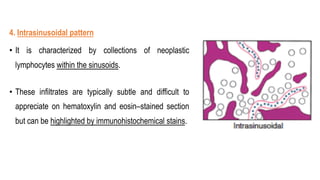




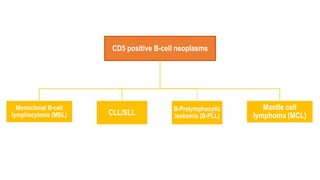
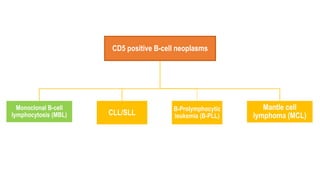
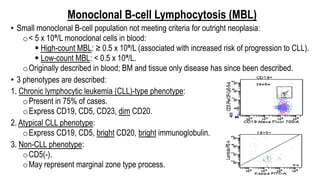
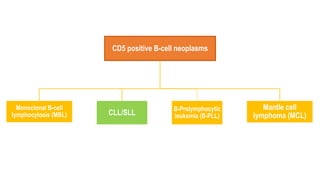
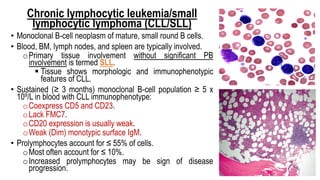

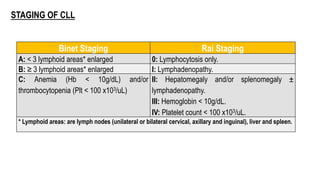
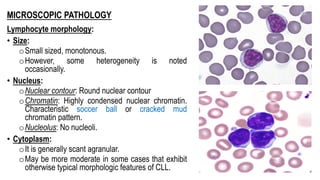
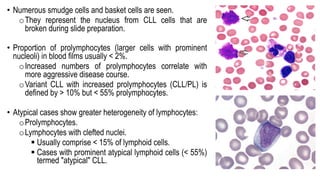
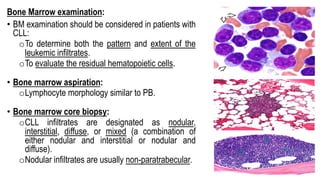
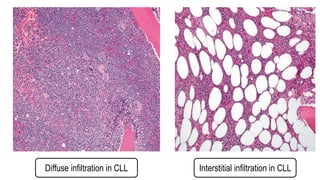
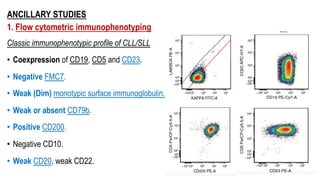
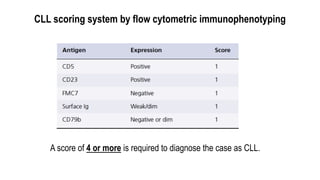
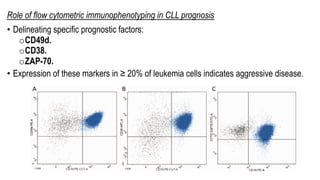
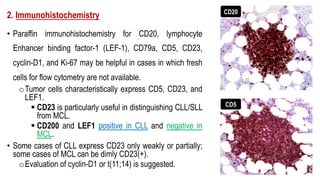

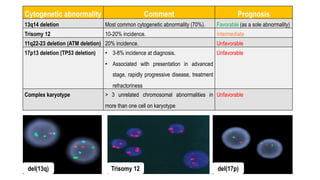

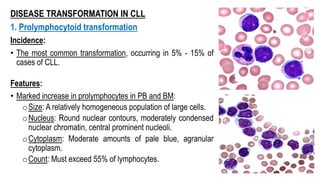
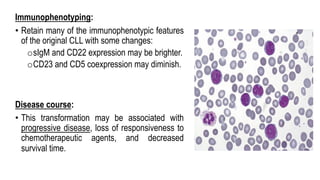
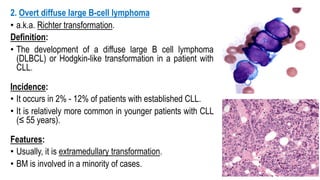
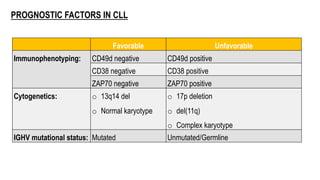
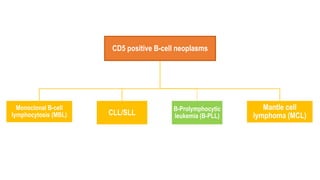


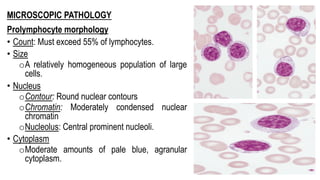
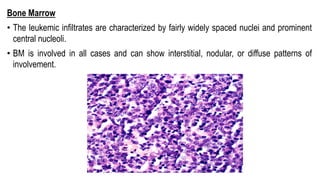
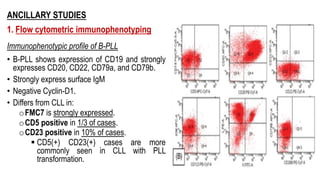

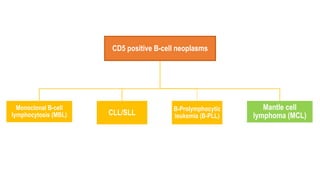
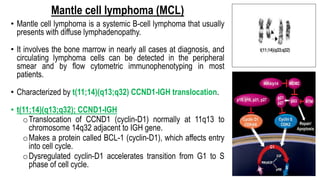

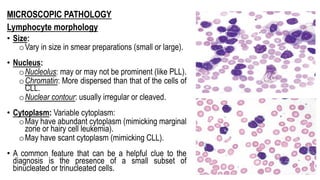
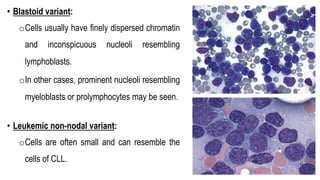
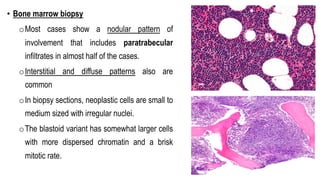
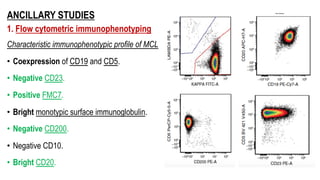

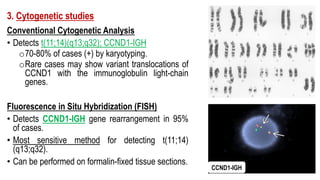
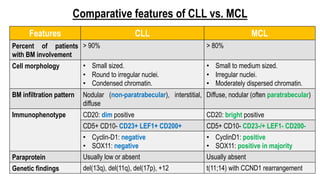
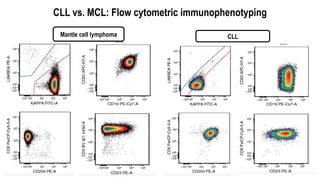

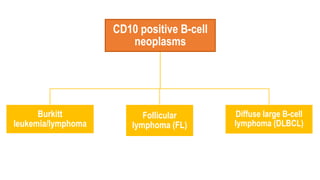



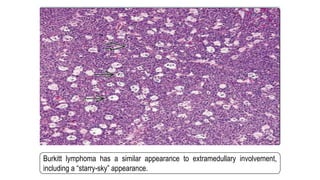
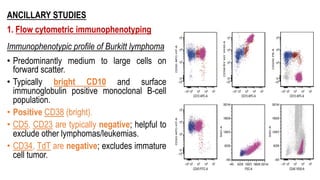
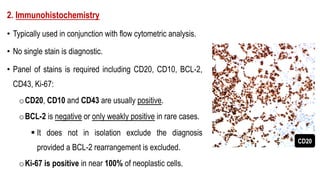

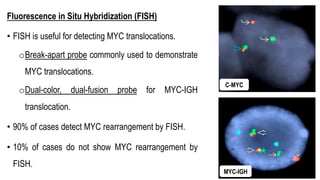
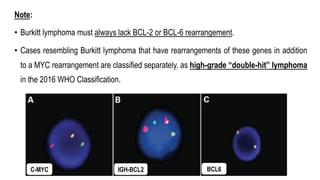
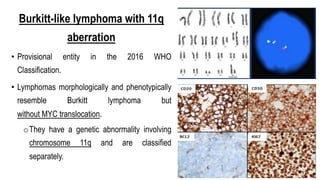
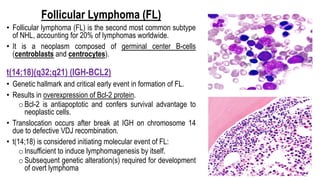

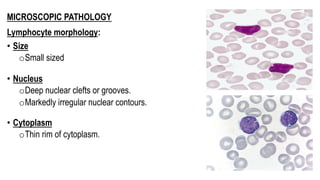
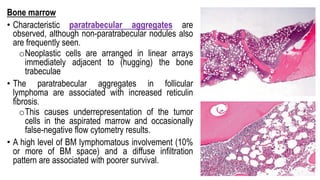
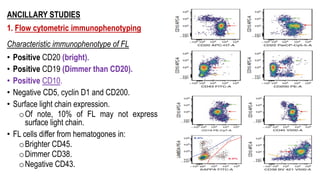
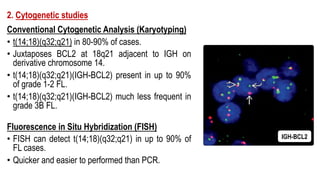
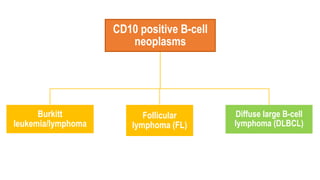
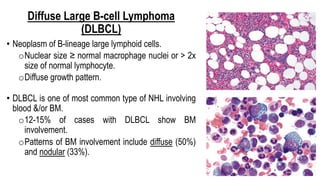


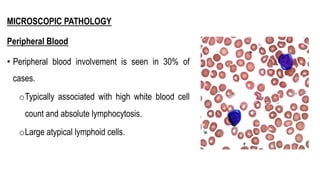
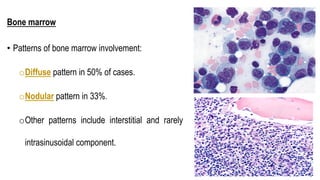


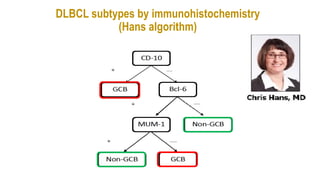
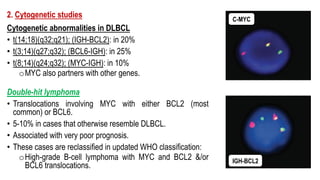


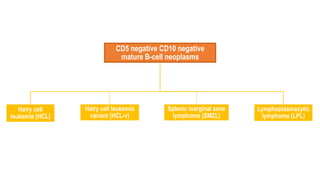
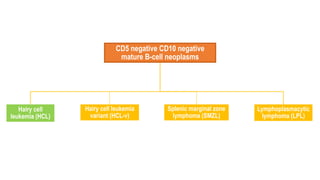
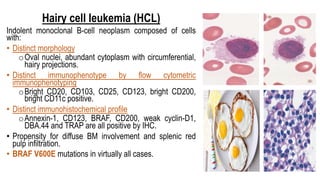

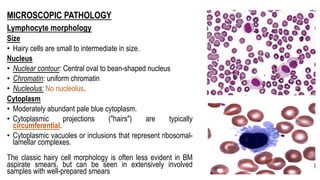
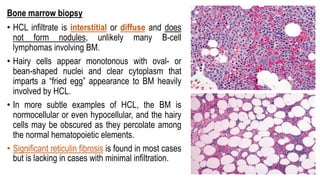
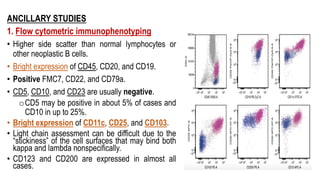

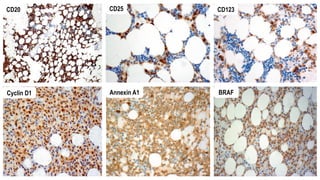
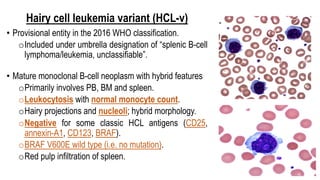
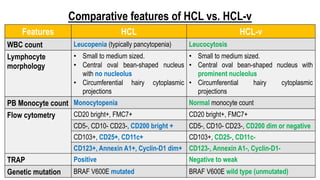
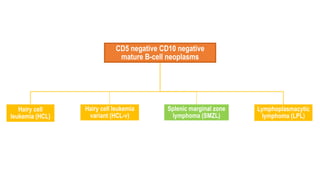
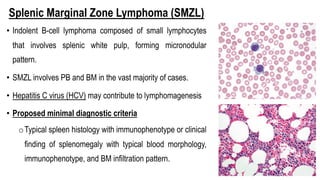

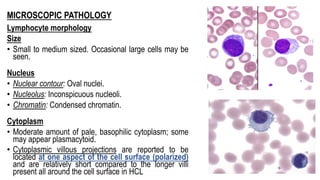
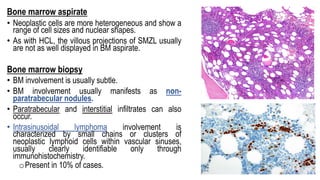
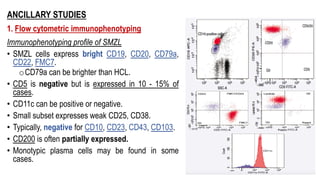

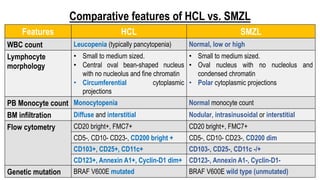
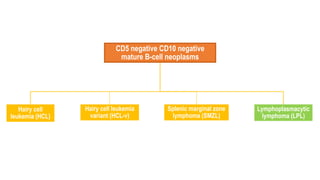
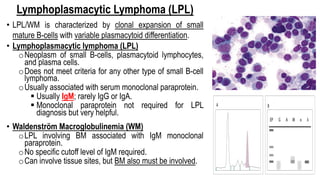

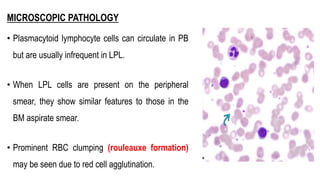
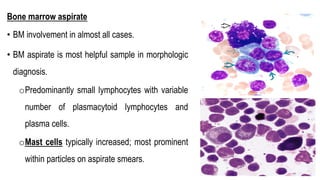
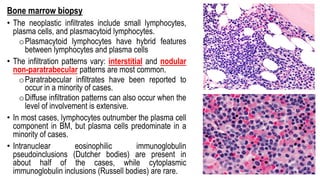
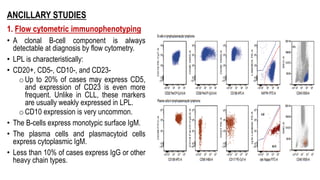
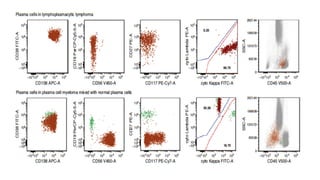

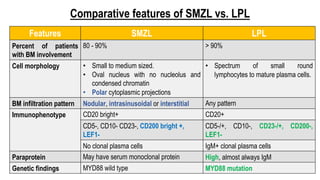
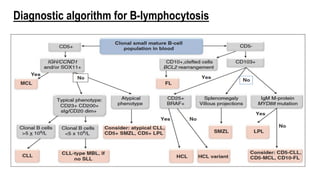

The document provides an overview of mature B-cell lymphomas and leukemias that involve the bone marrow and peripheral blood. It discusses several CD5 positive B-cell neoplasms including monoclonal B-cell lymphocytosis (MBL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), and B-cell prolymphocytic leukemia (B-PLL). CLL/SLL is characterized by small, monotonous lymphocytes and involves the blood, bone marrow, lymph nodes, and spleen. Prognostic factors in CLL/SLL include cytogenetic abnormalities, IGHV mutational status, and expression of cell surface markers. Rarely











































































