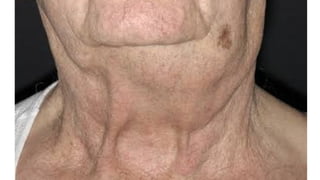
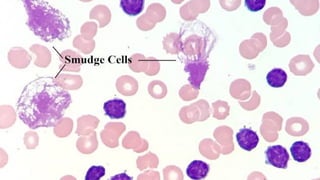
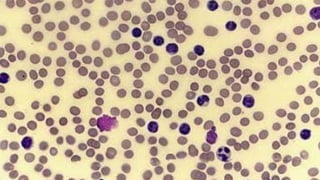
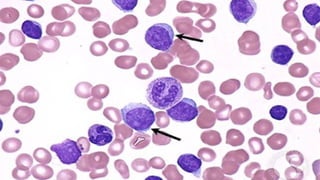
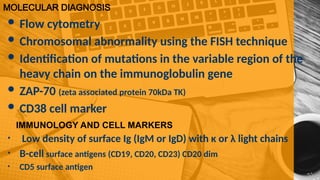
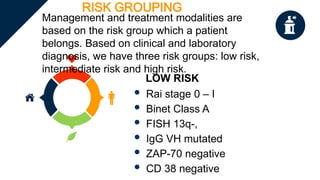
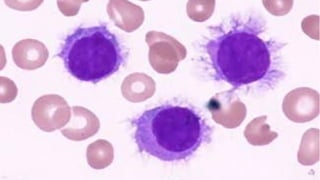
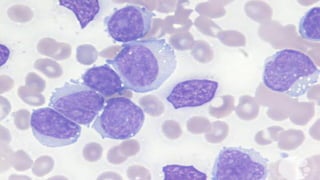
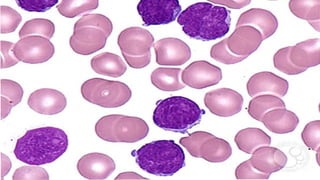
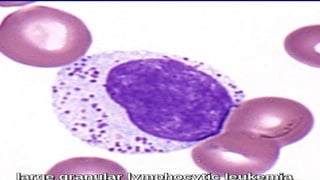
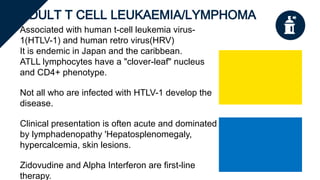
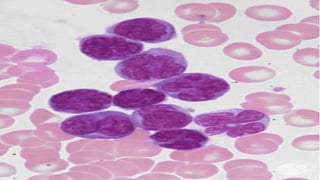
The document provides a comprehensive overview of chronic lymphocytic leukemia (CLL), detailing its clinical features, diagnostic criteria, staging classifications, treatment modalities, and prognosis. Key points include asymptomatic early stages, lymphocytosis, and the importance of risk grouping for management. The document also discusses various treatment options, including chemotherapy and newer targeted therapies, while highlighting other types of related leukemias.