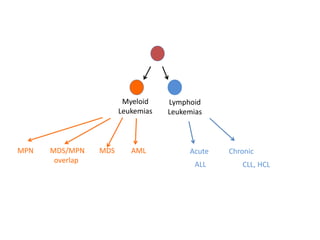
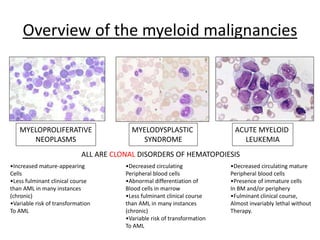
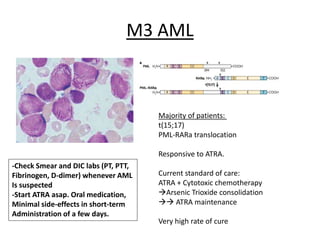
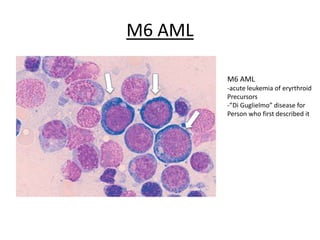
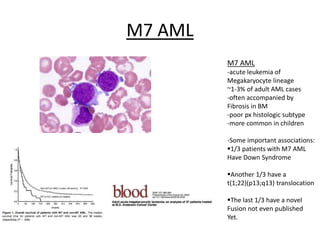
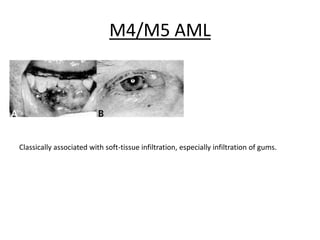
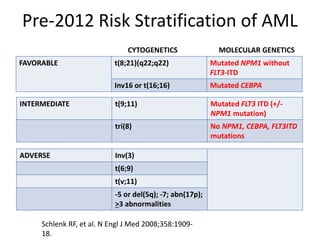
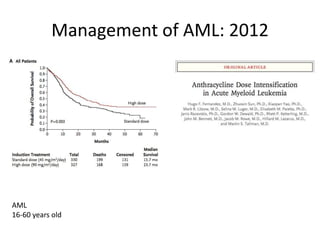
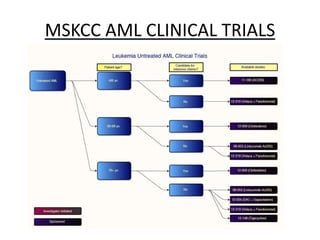
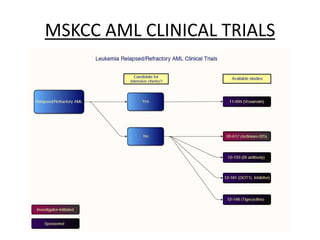
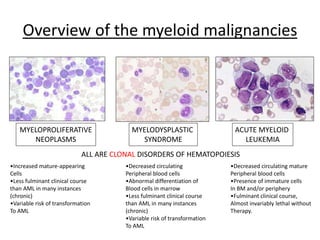
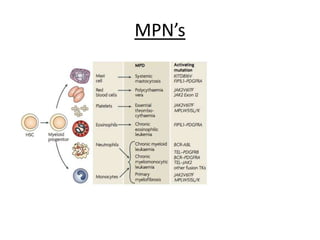
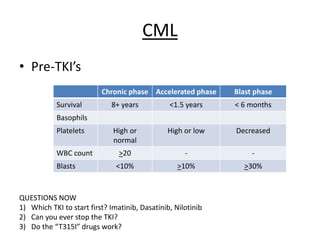
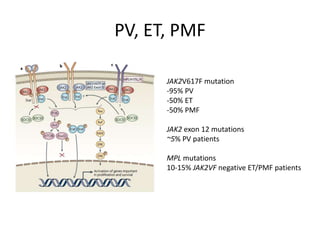
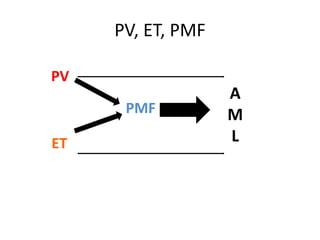
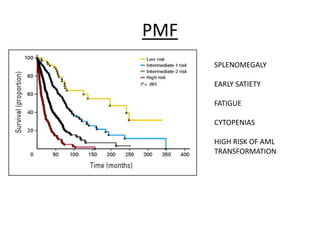
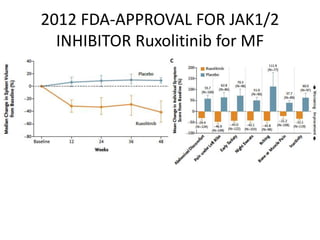
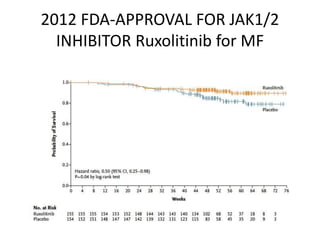
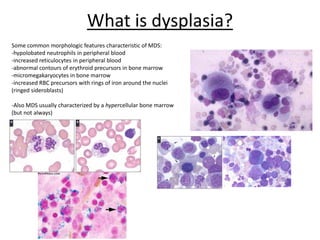
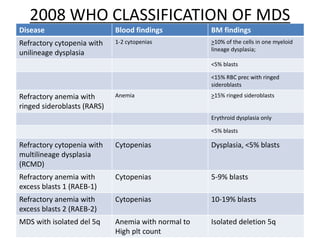
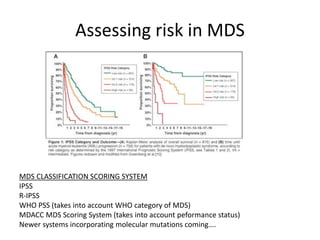
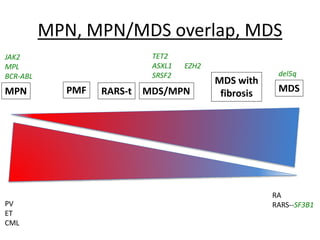
This document provides an overview of the myeloid malignancies, including myeloproliferative neoplasms (MPNs), myelodysplastic syndrome (MDS), and acute myeloid leukemia (AML). It describes key characteristics of each condition, such as increased mature cells in MPNs, decreased blood cells in MDS, and presence of immature cells in AML. Diagnostic criteria and classification systems for AML, including the 2008 WHO classification, are reviewed. Risk stratification in AML and standard treatment approaches are also summarized. Two clinical cases are then presented and discussed in detail.