Downloaded 454 times








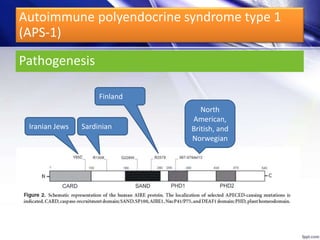
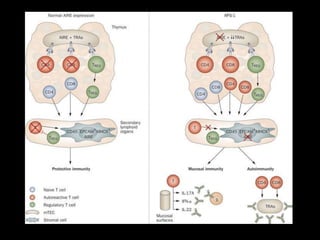
![Autoimmune polyendocrine syndrome type 1
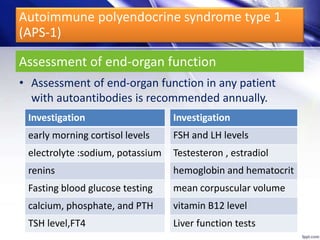
(APS-1)
• This condition is also termed as APECED autoimmune
polyendocrinopathy candidiasis ectodermal
dystrophy.
• Also called Whitaker's syndrome
• Due to a monogenetic mutation
• Males and females are equally affected
Dtsch Med Wochenschr. 2013 Feb;138(7):319-26; quiz 327-8. doi: 10.1055/s-
0032-1327355. Epub 2013 Feb 7.[Autoimmune polyglandular syndromes].
introduction](https://image.slidesharecdn.com/autoimmunepolyglandularsyndromes-140427032052-phpapp01/85/Autoimmune-polyglandular-syndromes-9-320.jpg)


































![• APS-2 is the most common autoimmune
polyendocrine syndrome.
• incidence of 1:20 000
• more common in females than in males.
• onset in adulthood.
• particularly during the third or fourth decades.
Dtsch Med Wochenschr. 2013 Feb;138(7):319-26; quiz 327-8. doi: 10.1055/s-0032-1327355. Epub 2013 Feb
7.[Autoimmune polyglandular syndromes].
Autoimmune polyendocrine syndrome type 2
(APS-2)](https://image.slidesharecdn.com/autoimmunepolyglandularsyndromes-140427032052-phpapp01/85/Autoimmune-polyglandular-syndromes-62-320.jpg)























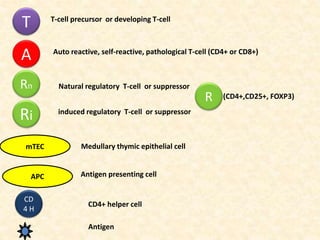
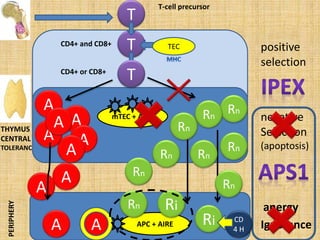
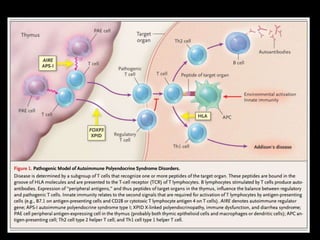
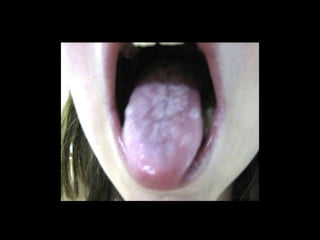
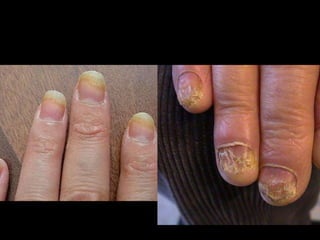
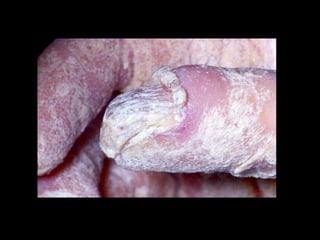
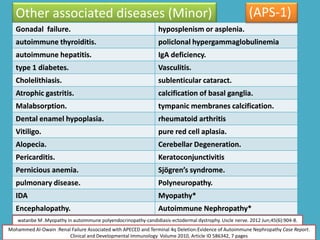
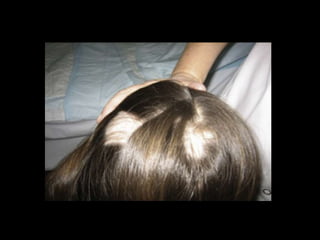
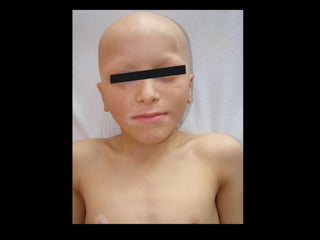
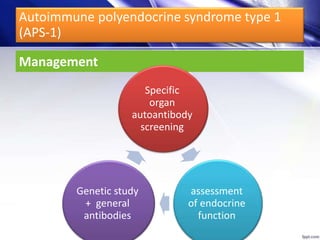
Dr. Yassin analyzed a 3-year old girl referred for hypoparathyroidism. Testing found she had a mutation in the AIRE gene, confirming autoimmune polyendocrine syndrome type 1 (APS-1). APS-1 is caused by AIRE gene mutations and results in autoimmunity against multiple endocrine organs. It commonly presents as chronic mucocutaneous candidiasis in childhood, followed by hypoparathyroidism and Addison's disease. Management requires monitoring and treating each individual condition as it arises.











































































































