Download as DOC, PPTX
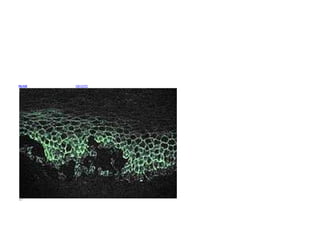
![Microscopic image of direct immunofluorescence
using an anti-IgG antibody. The tissue is skin from a
patient with Pemphigus vulgaris. Note the
intercellular IgG deposits in the epidermis and the
early intraepidermal vesicle caused by acantholysis.
Pemphigus ( /ˈpɛmfɪɡəs/ or /pɛmˈfaɪɡəs/) is a rare
group of blistering autoimmune diseases that affect
the skin and mucous membranes.[1]
In pemphigus, autoantibodies form against
desmoglein. Desmoglein forms the "glue" that
attaches adjacent epidermal cells via attachment](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-3-320.jpg)
![points called desmosomes. When autoantibodies
attack desmogleins, the cells become separated from
each other and the epidermis becomes "unglued", a
phenomenon called acantholysis. This causes blisters
that slough off and turn into sores. In some cases,
these blisters can cover a significant area of the skin.[2]
Originally, the cause of this disease was unknown,
and "pemphigus" was used to refer to any blistering
disease of the skin and mucosa. In 1964, a historic
paper that changed the understanding of pemphigus](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-4-320.jpg)
![was published.[3][4]
In 1971, an article investigating the
autoimmune nature of this disease was published.[5][6]
Contents
• 1 Types
• 2 Classification
• 3 Diagnosis
• 4 Treatment
•
Types](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-5-320.jpg)








![not considered part of the
Pemphigus group of diseases.[7]
Classification
Pemphigus is a group of
autoimmune blistering diseases that
may be classified into the following
types[8]
:](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-14-320.jpg)




![can affect the eyes and mucous
membrane of the oral cavity.
Intraorally it resembles the more
common diseases lichen planus and
mucous membrane pemphigoid.[9]
Definitive diagnosis requires
examination of a skin or mucous](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-19-320.jpg)




![important feature of the disease on
several WWW pages, but is not
mentioned in current Dermatology
textbooks nor do the terms
"Pemphigus" and "cANCA"
produce any hits on a PUBMED
search.[citation needed]](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-24-320.jpg)


![suppressant CellCept
(Mycophenolic acid) is among
those being used.[10]
Intravenous gamma globulin (IVIG)
may be useful in severe cases,
especially paraneoplastic
pemphigus. Mild cases sometimes](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-27-320.jpg)
![respond to the application of topical
steroids. Recently, Rituximab, an
anti-CD20 antibody, was found to
improve otherwise untreatable
severe cases of Pemphigus vulgaris.
[11][12]](https://image.slidesharecdn.com/autoimmunebullousdermatoses-130625085528-phpapp02/85/Autoimmune-bullous-dermatoses-28-320.jpg)















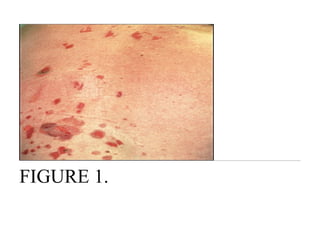
































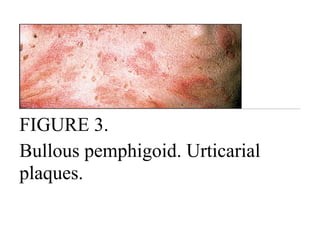


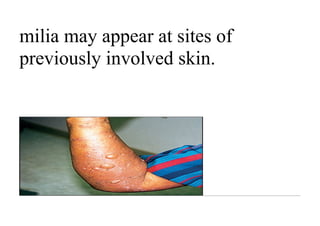












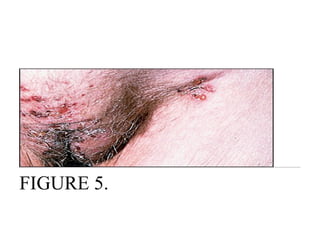

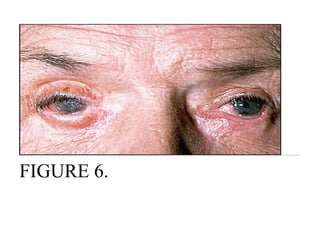

















































Pemphigus vulgaris is an autoimmune blistering disease that affects the skin and mucous membranes. It is caused by autoantibodies that attack desmoglein, a protein that binds epidermal cells together. This causes the epidermal cells to separate from each other (acantholysis) and form fragile blisters that rupture easily, leaving painful erosions. Pemphigus vulgaris is diagnosed through skin biopsy and detection of anti-desmoglein antibodies. Without treatment, it can be fatal due to infection; treatment involves high-dose corticosteroids and other immunosuppressants to control outbreaks and lessen side effects.



























































![[Chronic Multiple ulcers] O.Medicine lab](https://cdn.slidesharecdn.com/ss_thumbnails/o-250128123323-29e53ff5-thumbnail.jpg?width=640&height=640&fit=bounds)













































