Downloaded 37 times













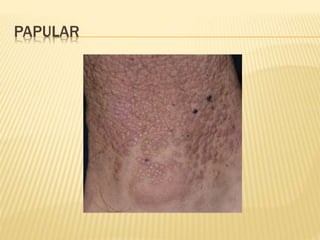
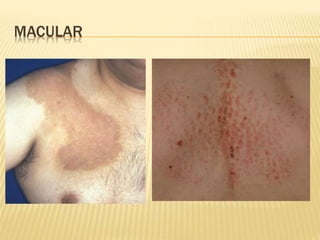
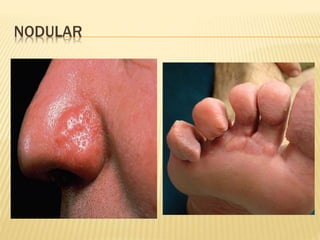


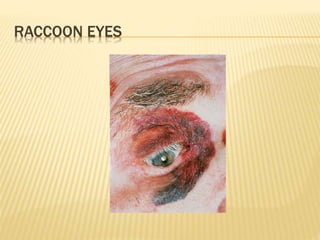
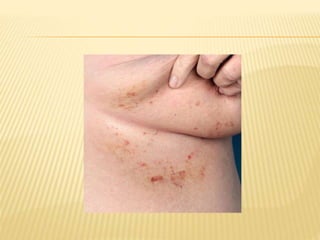
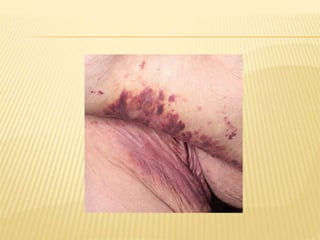
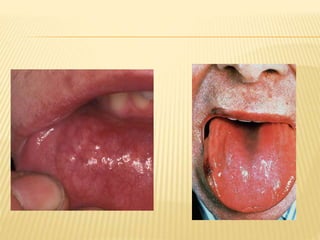


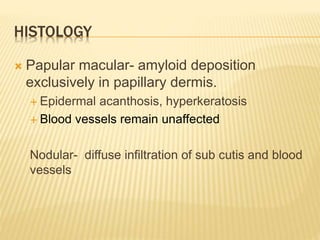

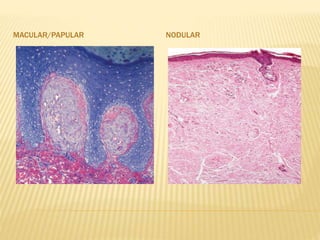

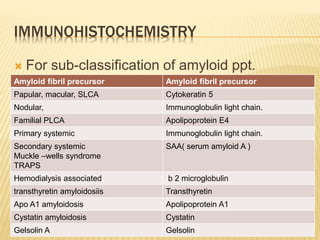
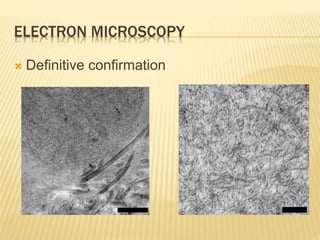
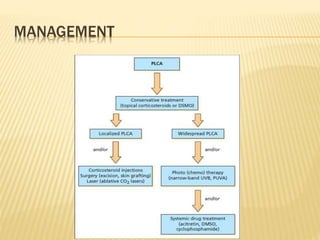

This document discusses cutaneous amyloidosis, which is a condition where abnormal protein deposits in the skin, changing its structure and function. It defines amyloidosis and describes the classification of primary localized, hereditary, and secondary systemic forms associated with other diseases. Key clinical features include yellowish macules and papules in localized forms, and petechiae, ecchymosis, and bleeding in secondary forms. Investigations include histology with amyloid stains like Congo red, immunohistochemistry to identify the amyloid precursor protein, and electron microscopy for definitive confirmation.

















































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)


![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)





![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)








