Downloaded 11 times



![PARKINSONISM PLUS
• Progressive Supranuclear Palsy
• Multiple System Atrophy [(Shy-Dragger syn.), SND (MSA P),
OPCA (MSA C)]
• Corticobasal Degeneration
• Dementia with Lewy Body Disease](https://image.slidesharecdn.com/parksem-200302044257/75/Parkinsonism-syndromes-with-differential-diagnosis-5-2048.jpg)











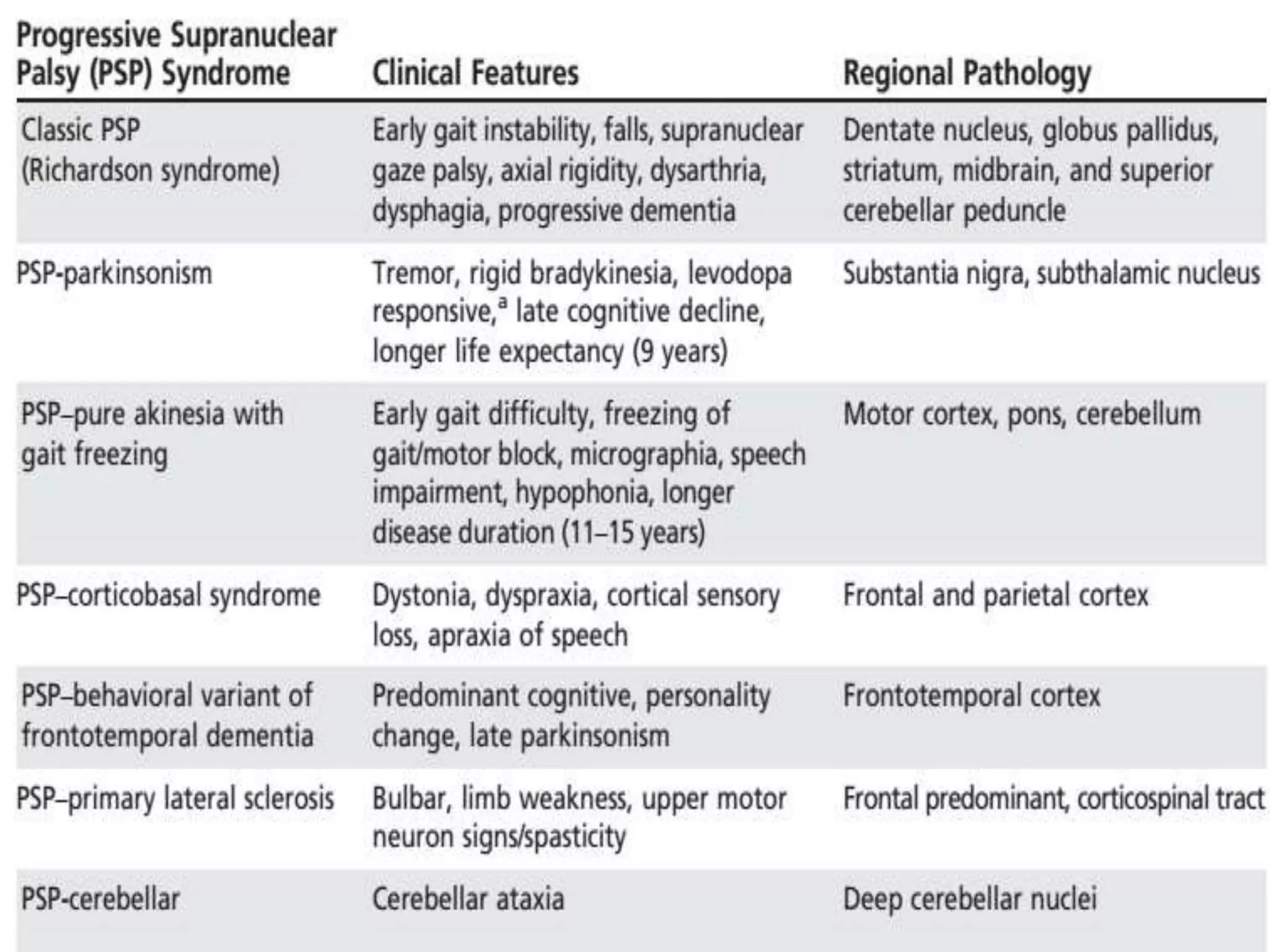




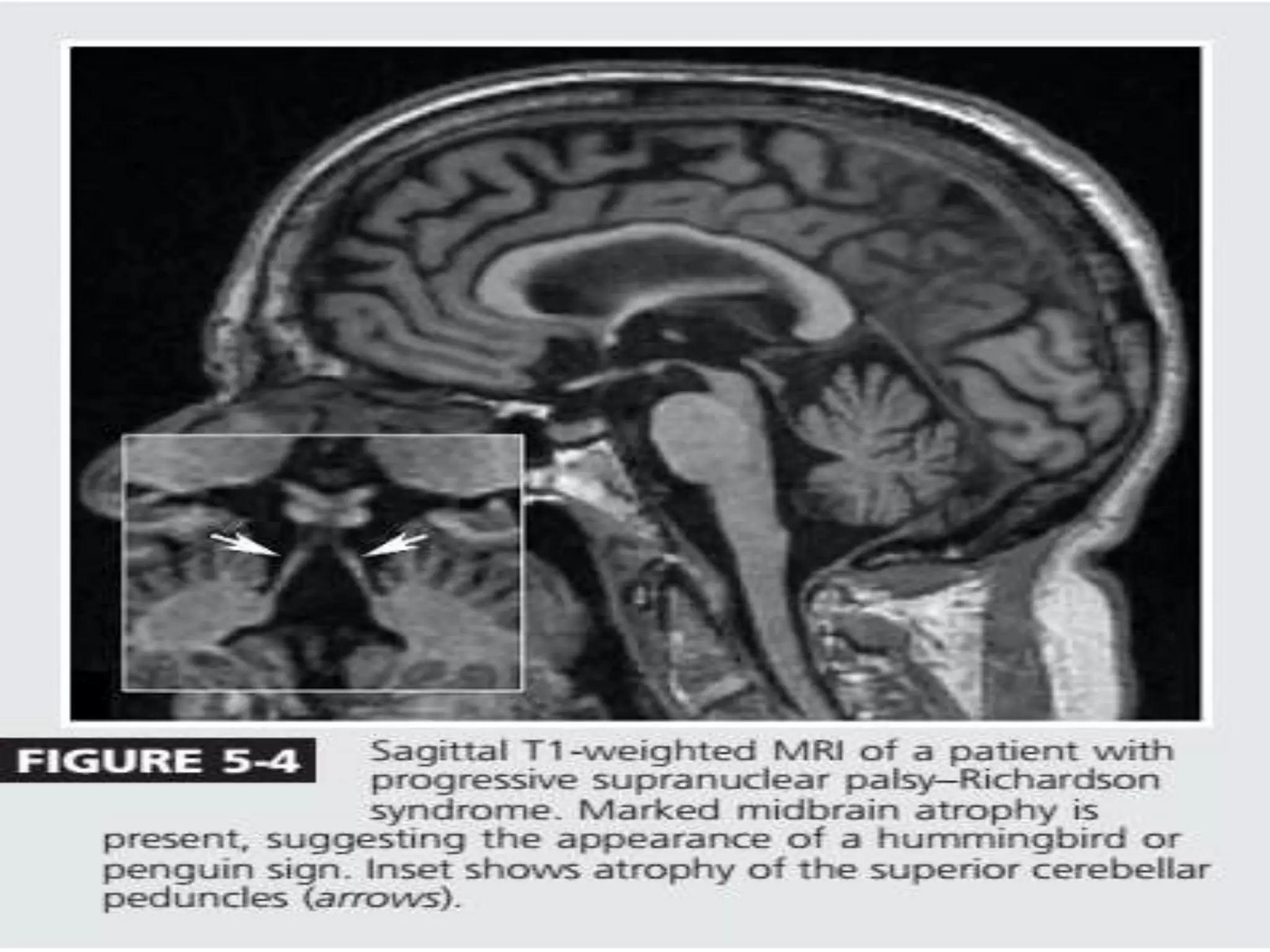
















































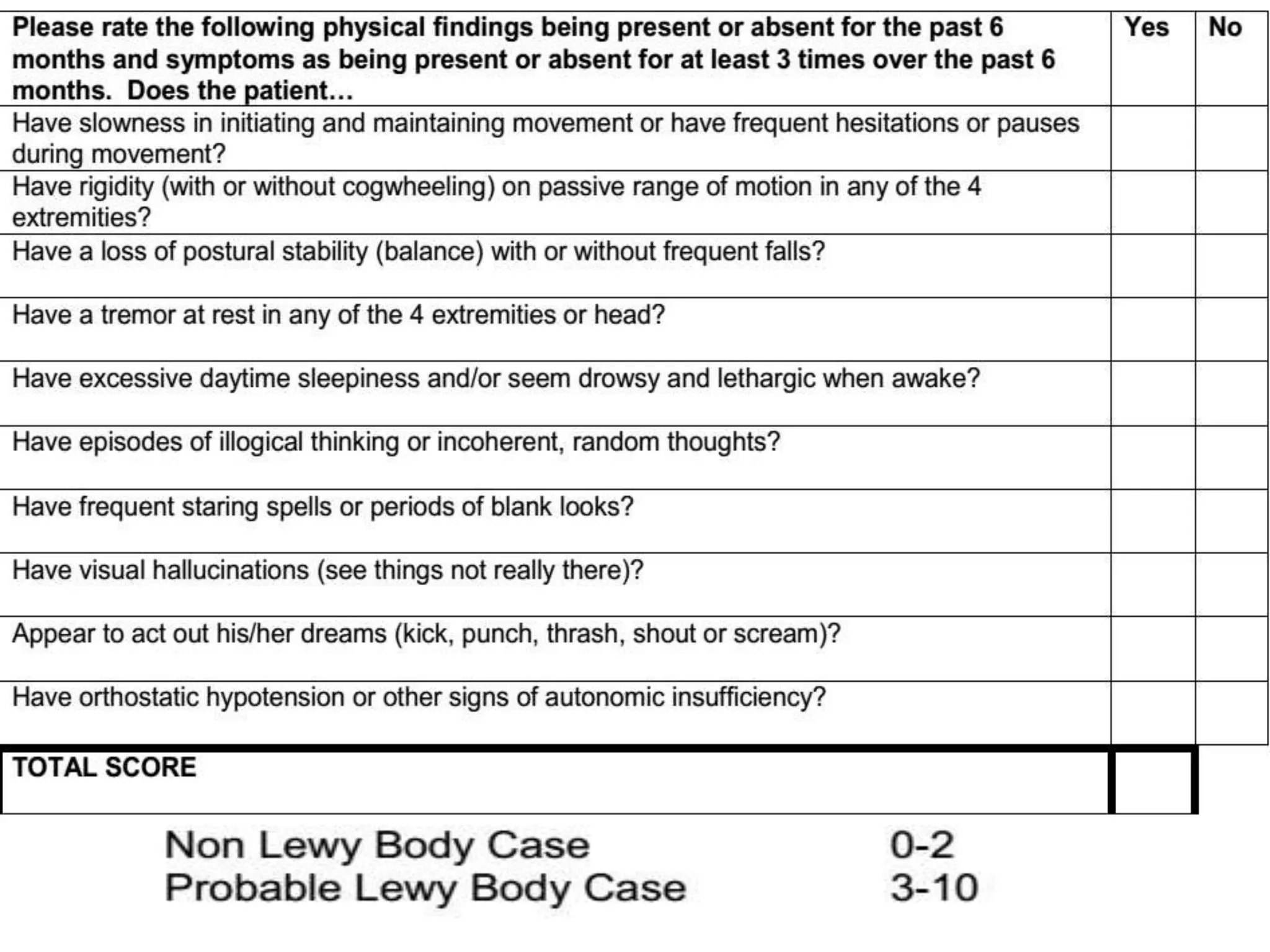



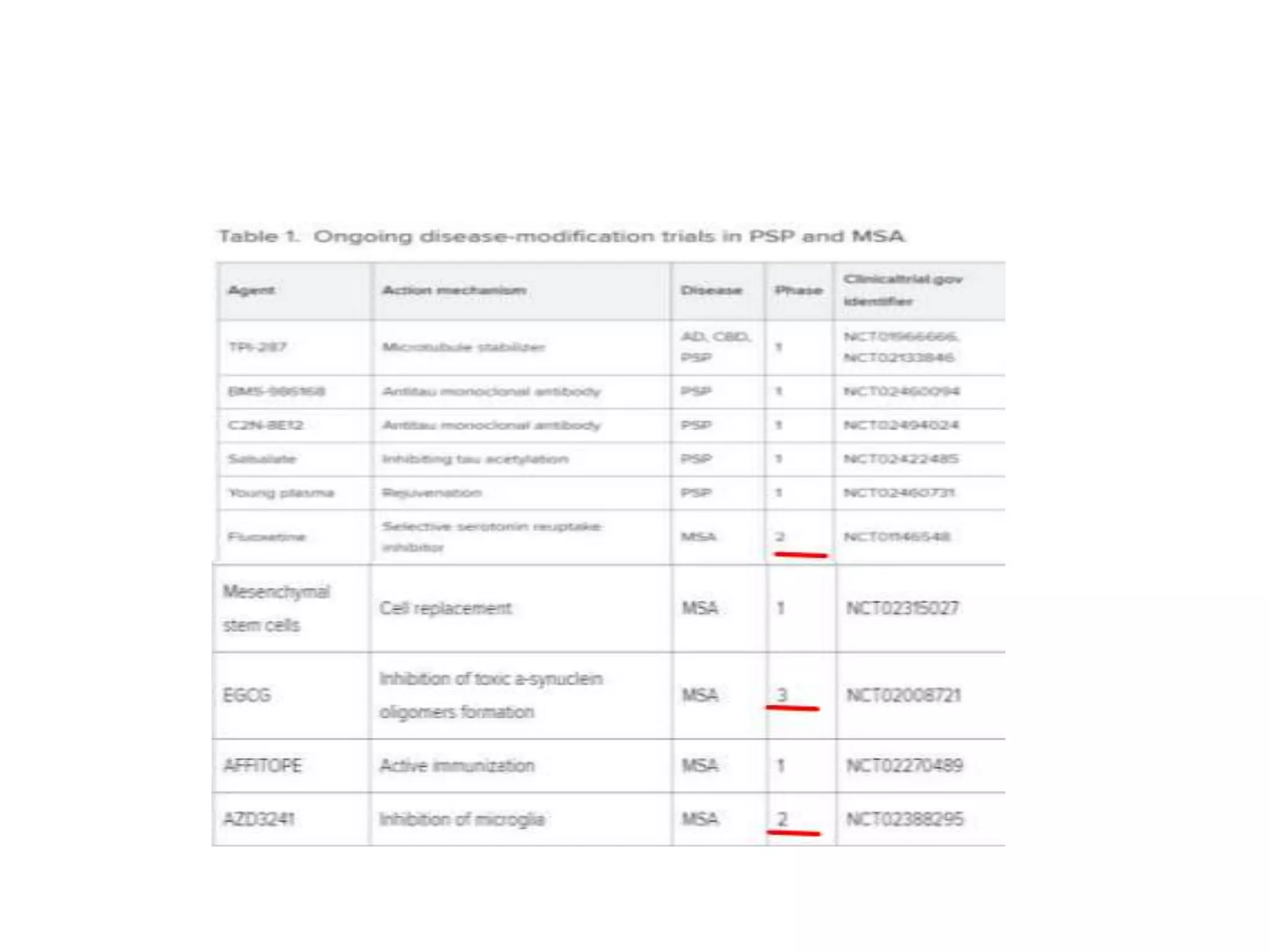




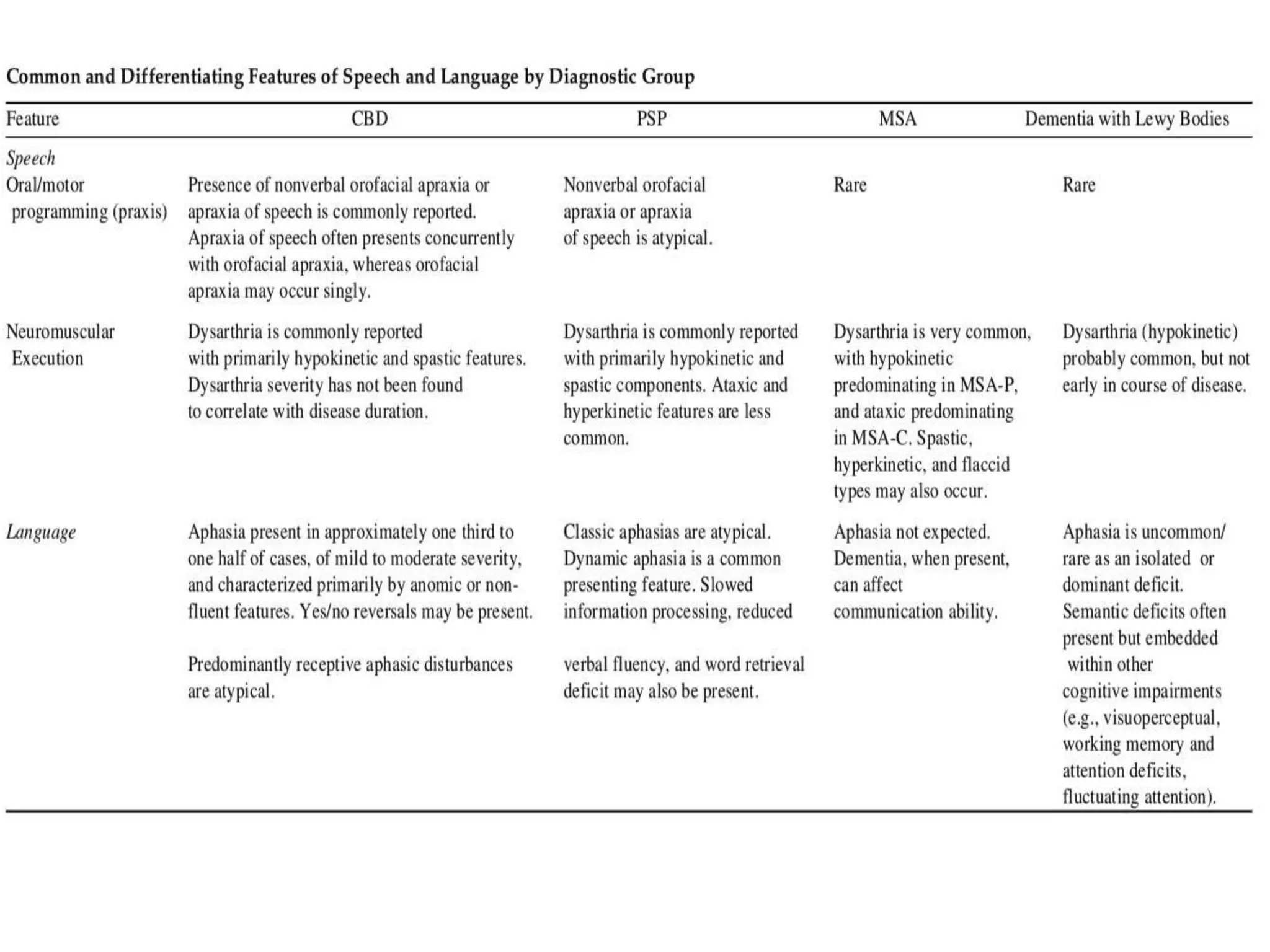


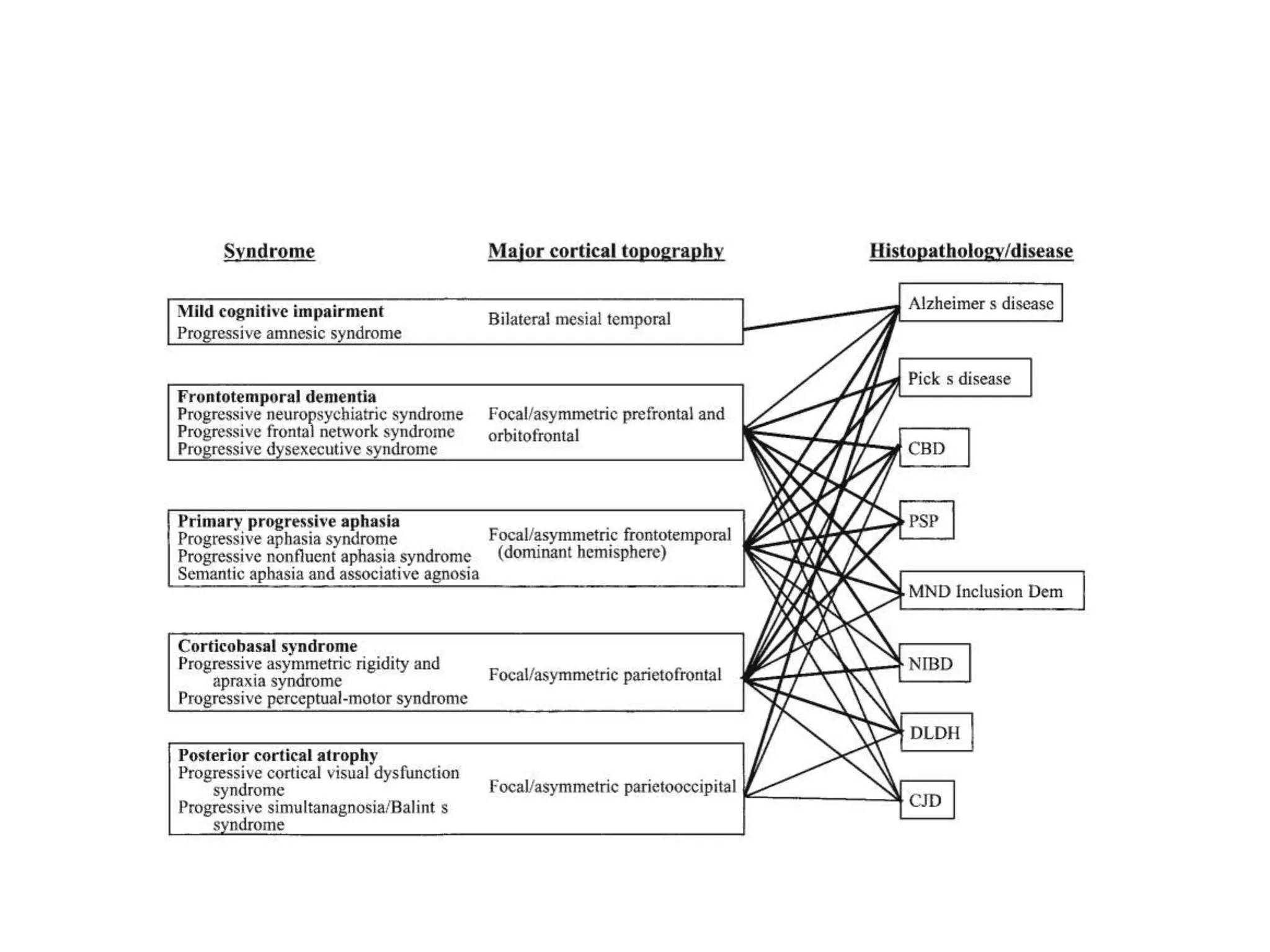






This document provides an overview of Parkinson plus syndromes, which are degenerative disorders that present with parkinsonism but have distinguishing features from idiopathic Parkinson's disease. It discusses several conditions in detail, including progressive supranuclear palsy (PSP), multiple system atrophy (MSA), and corticobasal degeneration (CBD). For each, it covers clinical features, diagnostic criteria, investigations, pathology, treatment approaches, and prognostic factors. The document aims to help clinicians differentiate these conditions from Parkinson's disease to guide appropriate management and prognostication.



































































