DEFINITION
Neurodegenerative disease isregression and progressive deterioration
of neurologic function with loss of speech, vision, hearing, or
locomotion, often associated with seizures, feeding difficulties, and
impairment of intellect.
3.
Neurodegenerative disordersare a group of heterogeneous diseases which result
from specific genetic, biochemical defect, chronic viral infection, toxic substances.
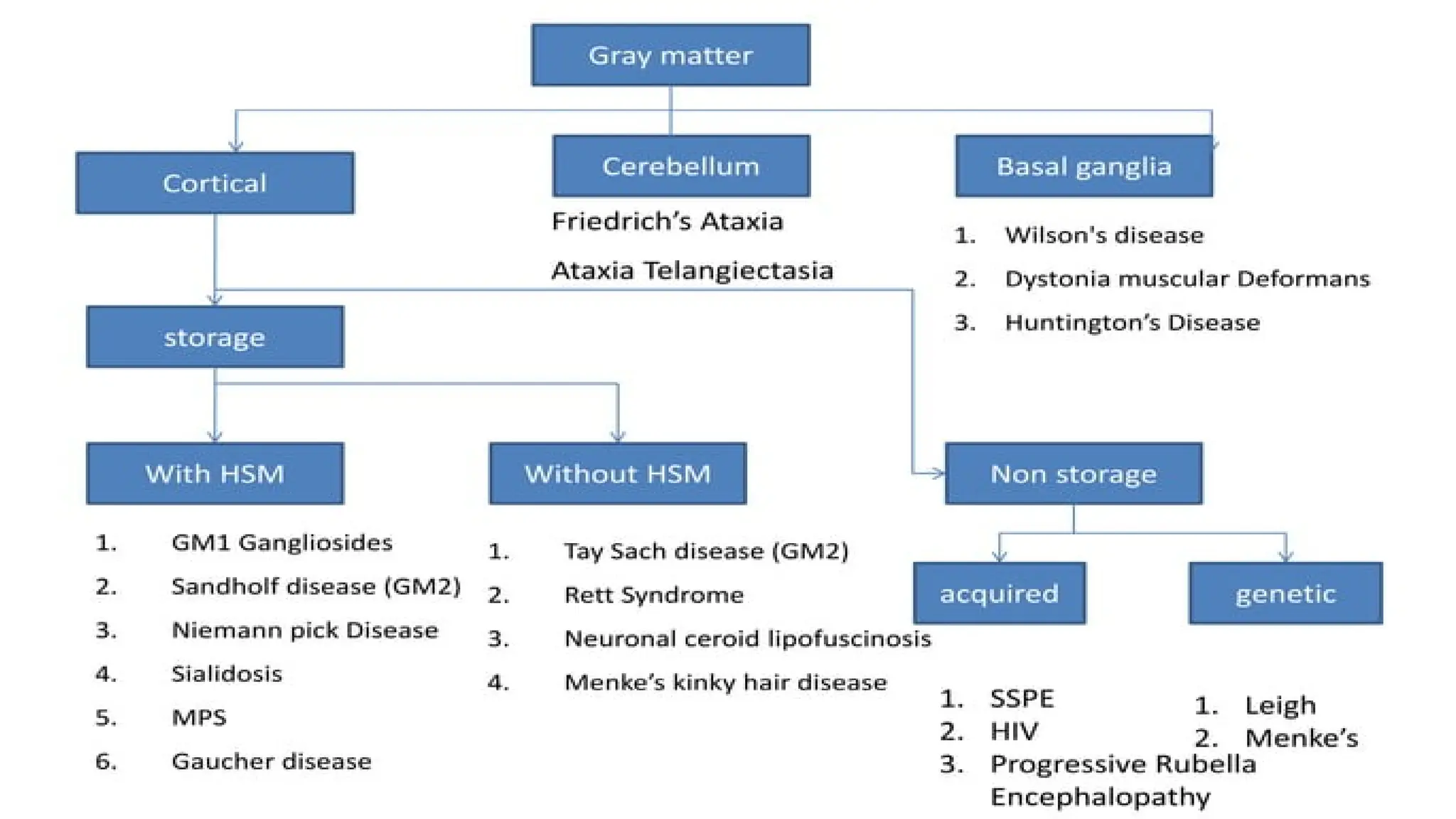
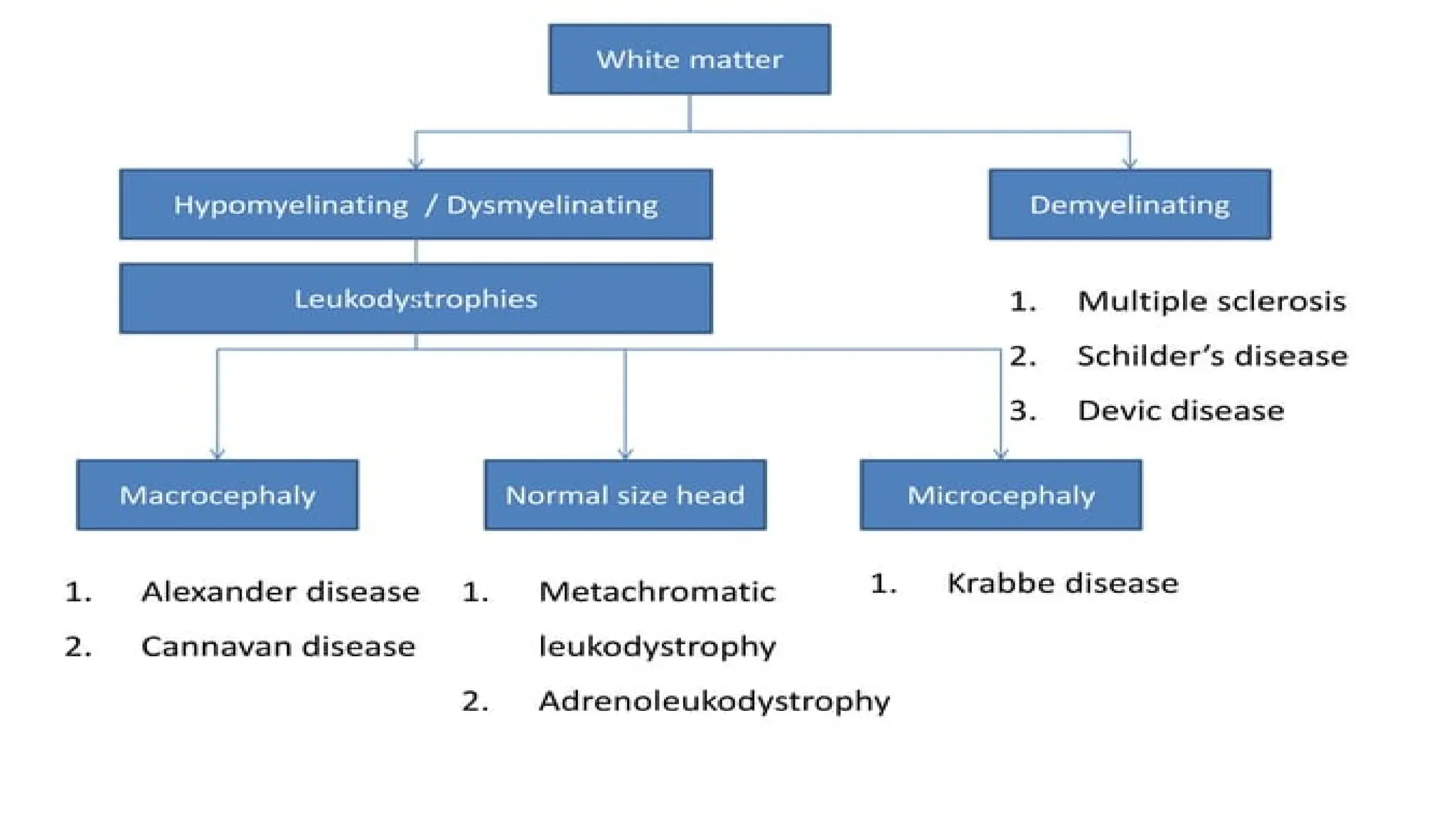
Involves both the gray matter and white matter.
The age of onset, rate of progression, and principal neurologic findings determine
whether the disease affects primarily the white or the gray matter
4.
Upper motorneuron signs and progressive spasticity are the
hallmarks of white matter disorders;
convulsions and intellectual and visual impairments that occur early
in the disease course are the hallmarks of gray matter disorders.
5.
Diffrentiating features Whitematter disorders Gray matter disorders
Age of onset Usually late (childhood) Usually early (Infancy)
Head size May have megalocephaly Usually microcephaly
Seizures Late ,rare Early , SEVERE
Cognitive functions Initialy normal Progressive dimentia
Peripheral neuropathy Early demylination Late , axonal loss
Spasticity Early , severe Late, progessive
Reflexes Absent(neuropathy),
Exaggeratted (long tract)
Normal or axaggerated
Cerebellar signs Early prominent Late
Fundal examinition May show optic atrophy Retinal degeneration
EEG Diffuse delta slowing Epileptic form discharge
EMG Slowed nerve conduction
velocity
Usually normal
ERG Normal Abnormal
6.
Classification of neurodegenerativebrain
disease:
Inherited
Gray matter
White matter
Both
Focal manifestations
Acquired
Infections Metabolic
8.
Metabolic causes
1. Chroniclead poisoning
2. Hypothyroidism
3. Vitamin B12 and E deficiency
4. Drugs (anticonvulsant)
10.
Classification According toAGE
Age at onset
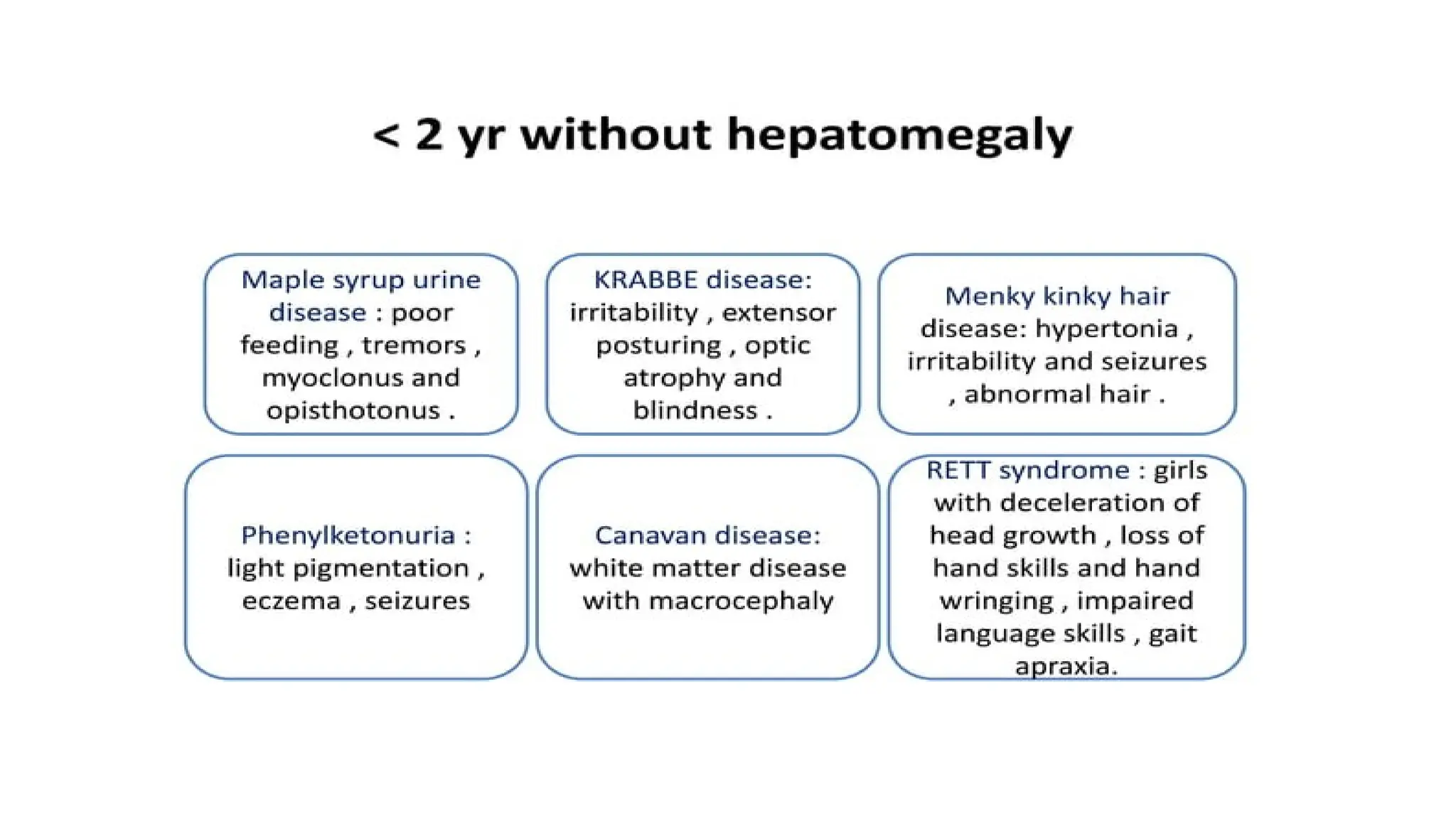
<2 year with Hepatomagly
<2 year without Hepatomagly
2 to 5 year
5 to 15 year
11.
AGE LESS THAN2 YEAR WITH HEPATOMEGALY
1. Fructose intolerance
2. Galactosemia
3. Glycogenosis (glycogen storage disease) types I-IV
4. Mucopolysaccharidosis types I and II
5. GM1 gangliosidosis
6. Niemann-Pick disease, infantile type
7. Zellweger syndrome
8. Gaucher disease (neuronopathic form)
9. Carbohydrate-deficient glycoprotein syndromes
15.
AGE LESS THAN2 YEAR WITHOUT HEPATOMAGLY
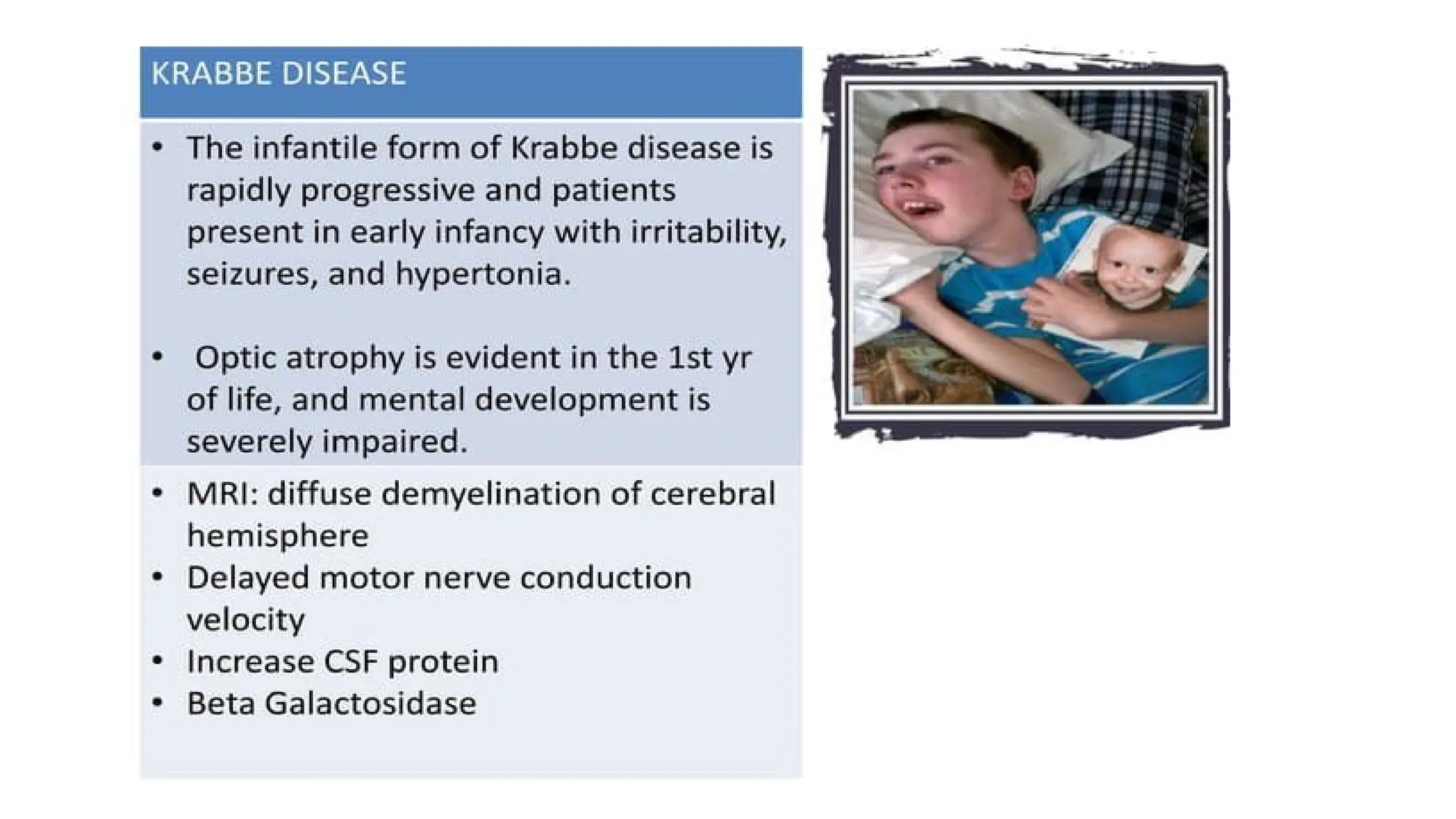
1. Krabbe disease
2. Rett syndrome
3. Maple syrup urine disease
4. Phenylketonuria
5. Menkes kinky hair disease
6. Tay-Sachs disease, GM2 gangliosidoses
7. Subacute necrotizing encephalopathy of Leigh disease
8. Canavan disease
9. Neurodegeneration with brain iron accumulation disease
19.
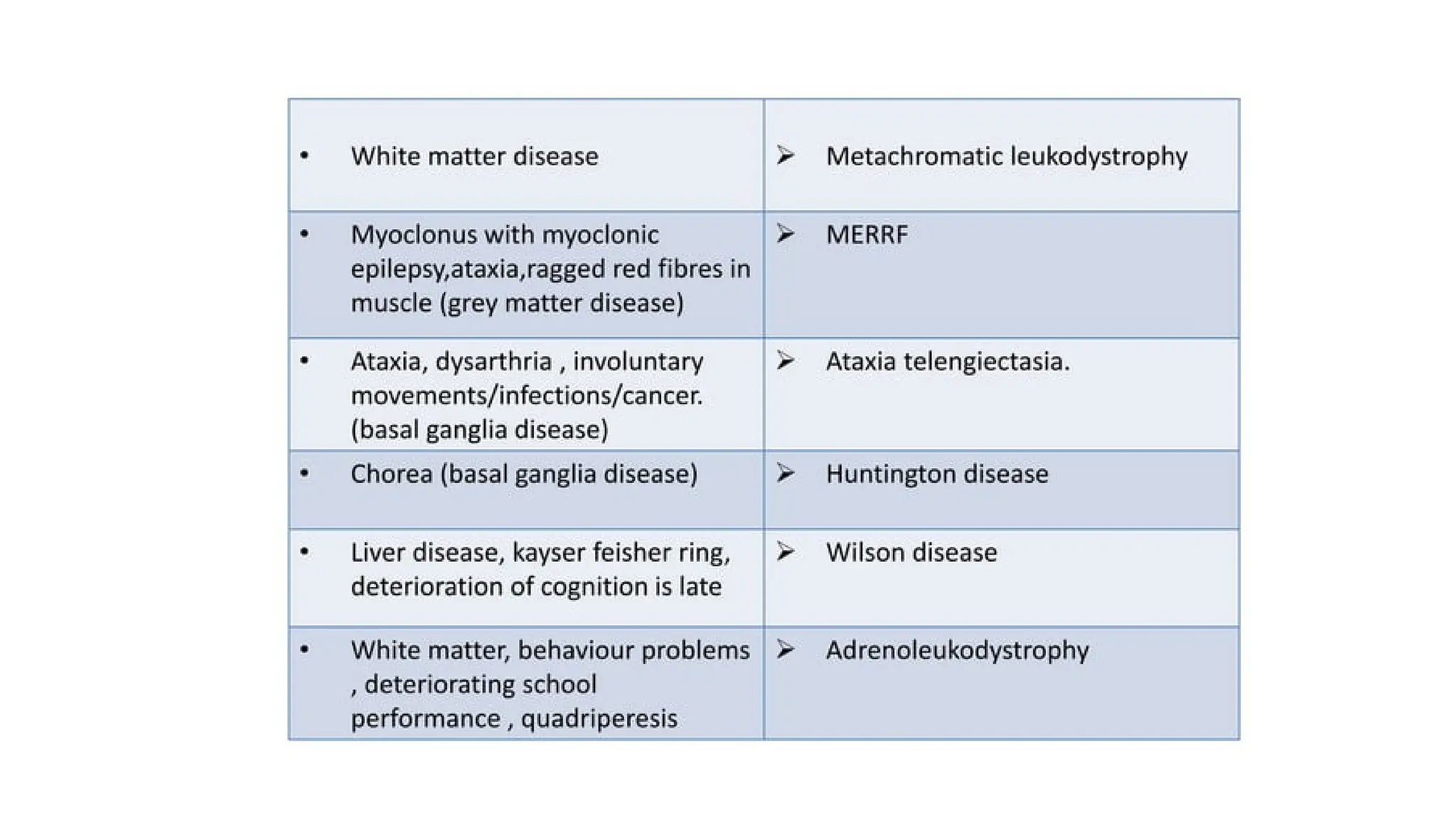
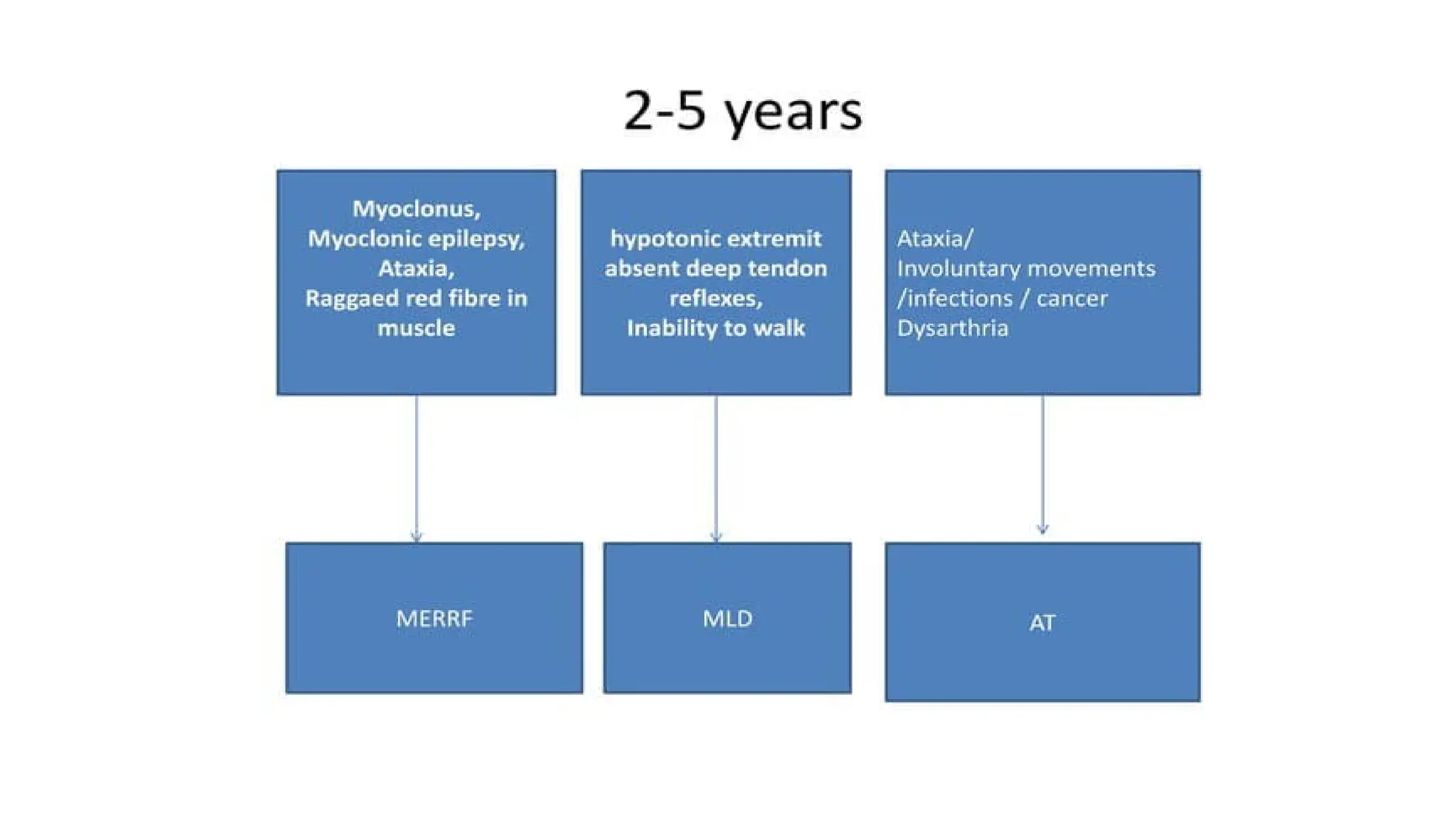
AGE 2 TO5 YEAR
1. Niemann-Pick disease types III and IV
2. Wilson disease
3. Neuronal ceroid lipofuscinosis
4. Mitochondrial encephalopathies
5. Ataxia–telangiectasia
6. Neurodegeneration with brain iron accumulation syndrome
7. Metachromatic leukodystrophy
8. X-LINKED Adrenoleukodystrophy
24.
Neuro Wilson Presentation
A complication of Wilson’s disease due to copper accumulation in the brain
Primary organ affected initially: Liver → Copper then deposits in the brain (basal
ganglia) and eyes (Kayser-Fleischer rings).
Kidney involvement: Can lead to Fanconi syndrome.
Neurological Symptoms:
Movement disorders: Dystonia Chorea Chorea - athetosis Tremors
Basal ganglia dysfunction leads to the "4Ds": 1. Dystonia 2. Drooling
3. Dysphagia (difficulty swallowing) 4. Dysarthria (slurred speech)
25.
MRI findings inNeuro Wilson
Dilatation of ventricles
Atrophy of cerebrum, cerebellum, and brainstem
↑ signal intensities in Basal ganglia, Mid-brain, Thalamus (Mnemonic: BMT)
These findings combined are called "PANDA Sign“ Another similar finding of "Eye
tiger sign" is present in Hallervorden- Spatz disease, which is also a DBD.
28.
AGE 5 TO15 YEAR
1. Adrenoleukodystrophy
2. Neuronal ceroid lipofuscinosis, juvenile and adult forms
3. Refsum disease
4. Sialidosis II, juvenile form
5. SSPE
29.
SUBACUTE SCLEROSING PANENCEPHALITIS
(SSPE)
SSPEis a white matter DBD and commonly presented in advanced neurology setups.
Cause is persistent infection with altered measles virus. This alteration is “Missing of M-Protein.”
Measles virus remains intracellular for many years (7-10 years) and then reactivates.
Measles virus stays within cells, and is thus saved from humoral immunity.
Younger age of acquired measles, more chances of developing SSPE due to immature immunity.
(Among SSPE patients, 50% had measles at < 2 yr age, and 75% have measles at < 4 yr age.)
31.
Investigations
1. Imaging:
2. Radiographs:
3.Urine and blood assay for plasma ammonia, blood lactate and pyruvate, plasma amino acids:
4. Specific investigations are done based on clues obtained from preceding investigations
5. Electrophysiological test
6. Histopathological and ultrastructural information from selected biopsies
A,Bone marrow: storage cells are seen in Niemann-Pick disease, Gaucher disease
B, Conjunctival, skin, rectal biopsy: Neuronal ceroid lipofuscinosis
C. Hair microscopy: Menkes Disease
8. Serology: HIV, SSPE
9. Urine copper, serum ceruloplasmin: Wilson disease
10. Urine MPS: mucopolysaccharidosis
11. Enzyme analysis: lysosomal storage disorders, biotinidase deficiency
12. Urine organic acids: organic acidemias
13. Very long-chain fatty acids (VLCFA) and plasmalogen levels: peroxisomal disorders
14. Mutation testing: this can be undertaken if the diagnosis is quite certain and the test is available
32.
NEURODEGENGRATIVE DISORDER DIGNOSTIC TEST
Canavan disease N - acetylaspartic acid (urine)
Alexander disease β-crystallin (CSF)
Krabbe leukodystrophy β-galactosidase (leukocytes/fibroblasts)
Metachromatic leukodystrophy Arylsulfatase A (leukocytes/fibroblasts)
Adrenoleukodystrophy Very long chain fatty acids (VLCFA)
Mucopolysaccharidosis Mucopolysaccharides (urine)
Sialidosis (Type 1) α-neuraminidase (leukocytes/fibroblasts)
Neuronal ceroid lipofuscinosis Skin, conjunctival, or rectal biopsy
Mitochondrial encephalopathies Lactate (CSF/blood), muscle biopsy
Refsum disease Phytanic acid (blood)
Lesch-Nyhan disease Hyperuricuria and hyperuricemia
Spinal muscular atrophy Muscle biopsy, DNA studies (blood)
Friedreich ataxia DNA studies (blood)
Wilson disease Urine copper, serum copper, and
ceruloplasmin
Menkes kinky hair syndrome Serum copper and ceruloplasmin
33.
Management
Firstly, nevermiss out a treatable cause of neuroregression like hydrocephalus, HIV infection,
hypothyroidism, lead toxicity, etc. It must be remembered that many NDD are amenable to treatment.
Supportive Measures
It is never wise or correct to say that the disease is “untreatable.”
Something can almost always be done to help the child. Supportive treatment may add significantly to
the quality of life of a child with a neurodegenerative disorder.
Measures to reduce spasticity, control seizures, control pain, improve nutrition, prevent constipation,
prevent bed sores, and enhance mobility all contribute to the quality of life of the patient and indirectly
to the quality of life of the parents and any unaffected siblings.
Supportive: The treatable complications
feeding difficulties, gastroesophageal refluxspasticity, drooling skeletal deformities,
And recurrent chest infections epilepsy, sleep disorder, behavioral symptoms.
34.
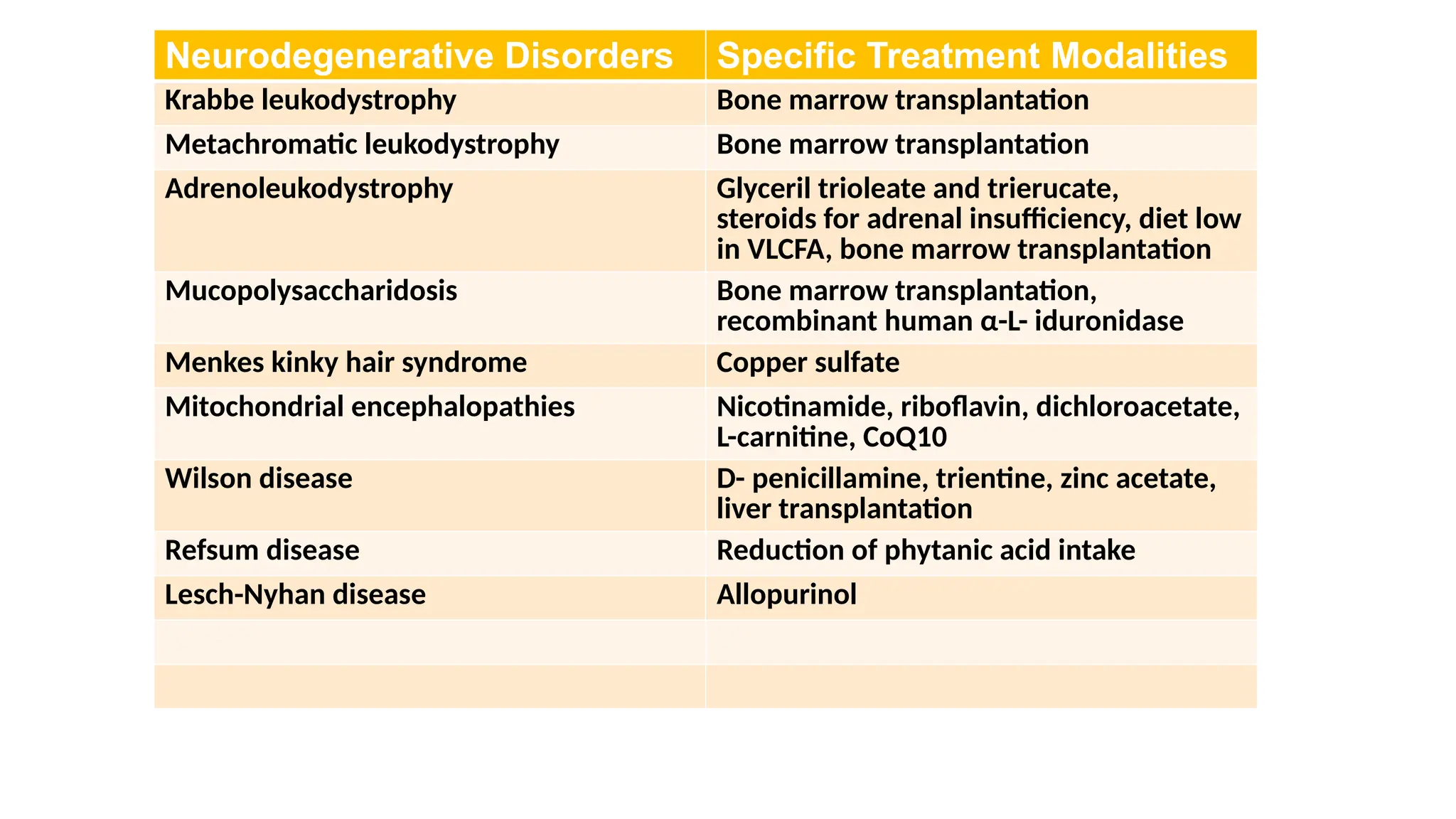
Neurodegenerative Disorders SpecificTreatment Modalities
Krabbe leukodystrophy Bone marrow transplantation
Metachromatic leukodystrophy Bone marrow transplantation
Adrenoleukodystrophy Glyceril trioleate and trierucate,
steroids for adrenal insufficiency, diet low
in VLCFA, bone marrow transplantation
Mucopolysaccharidosis Bone marrow transplantation,
recombinant human α-L- iduronidase
Menkes kinky hair syndrome Copper sulfate
Mitochondrial encephalopathies Nicotinamide, riboflavin, dichloroacetate,
L-carnitine, CoQ10
Wilson disease D- penicillamine, trientine, zinc acetate,
liver transplantation
Refsum disease Reduction of phytanic acid intake
Lesch-Nyhan disease Allopurinol
35.
Prognosis
The prognosisin NDD depends on the underlying disorder. In general, an earlier onset of disease
predicts a poorer outcome. However, several late-onset diseases can also rapidly progress, e.g.,
adrenoleukodystrophy ages.
Always keep the treatable causes in mind and exclude them systematically, based on the clinical
presentation.
While managing these children, always focus on issues that make the child's life comfortable, e.g.,
control seizures, spasticity, adequate nutrition, and skin care, etc