Downloaded 131 times

















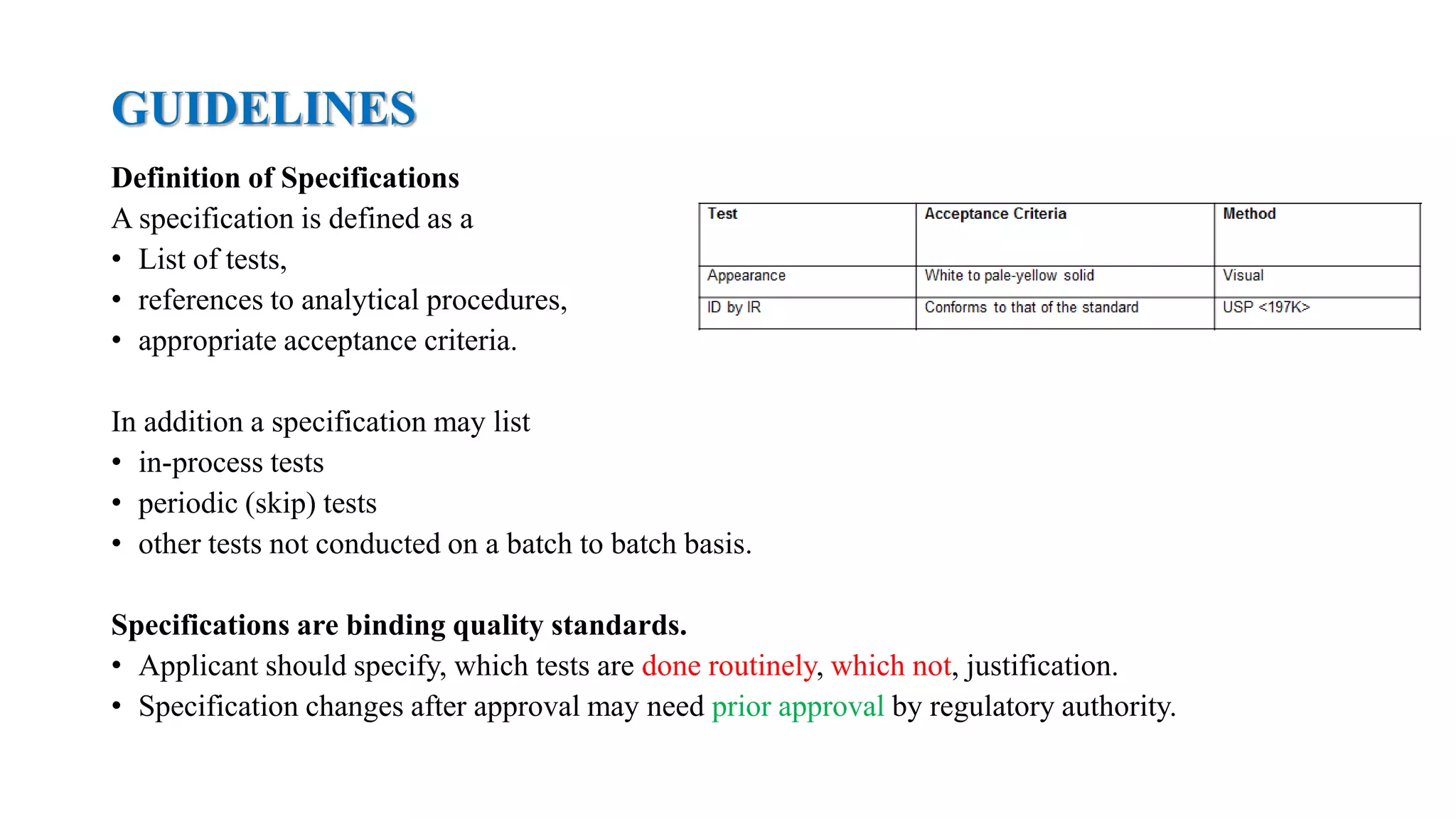






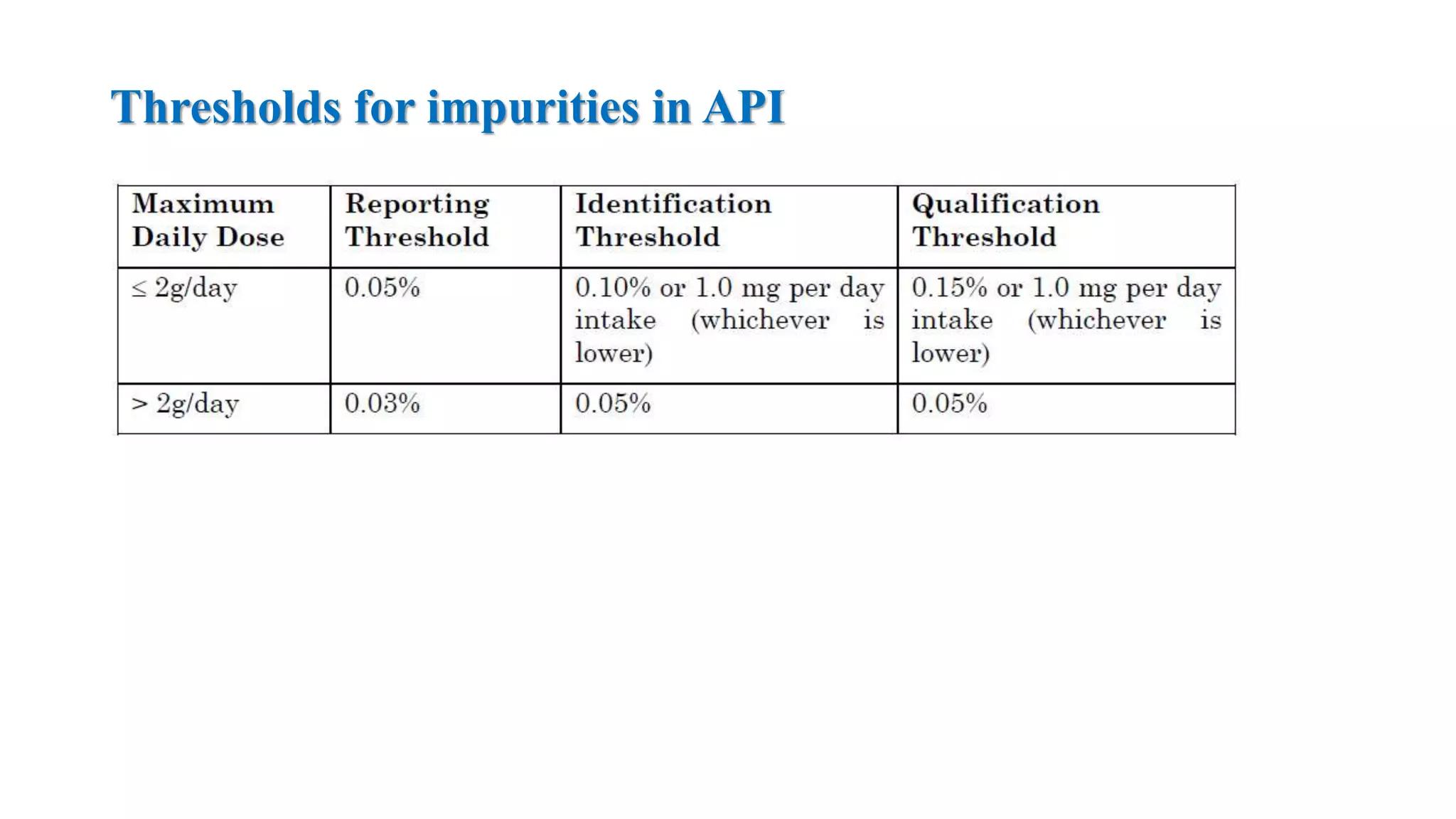


















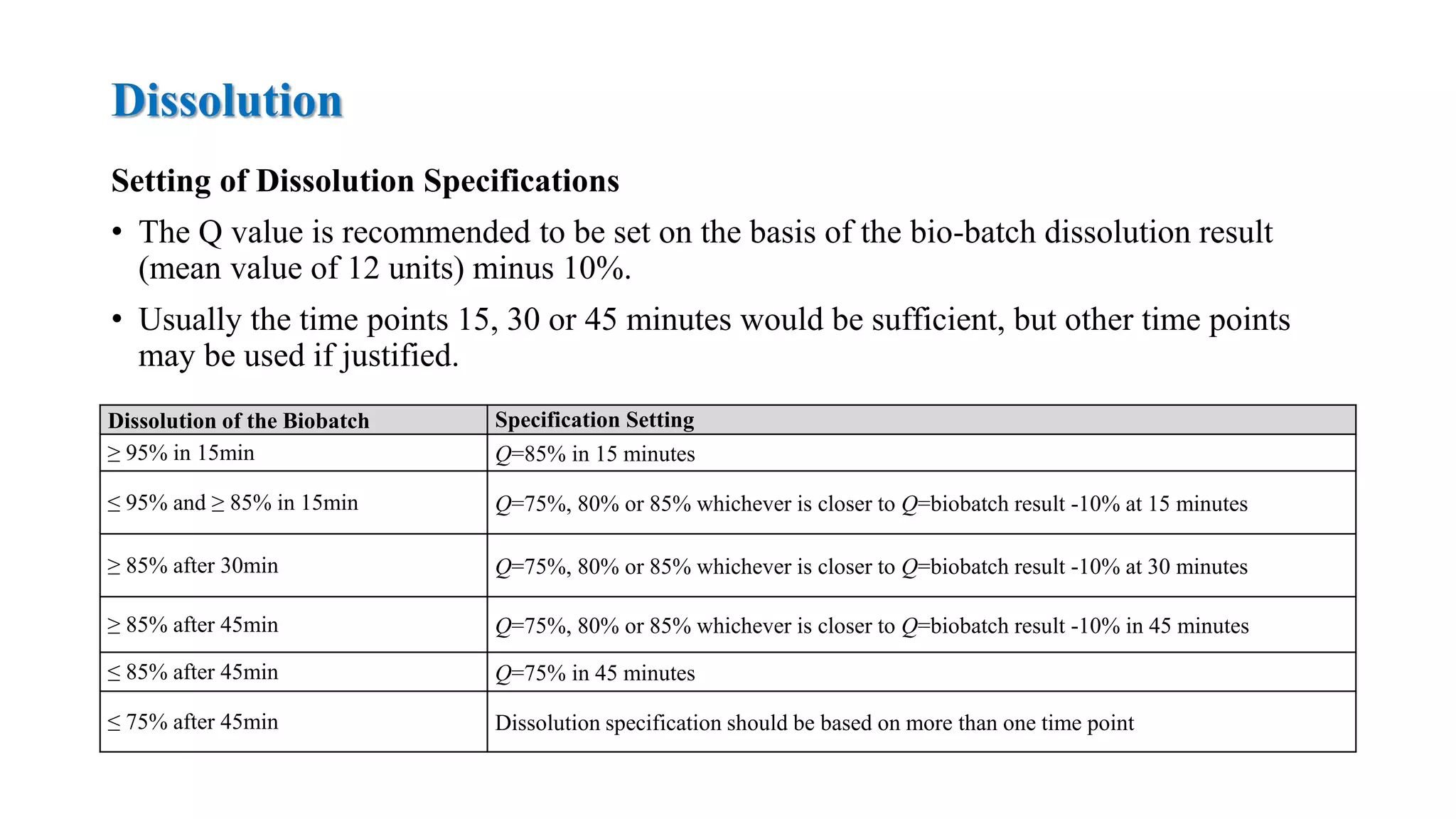


















The document provides guidelines on specifications for new drug substances and products. It defines specifications and outlines universal tests that should be included for both drug substances and products, such as description, identification, assay, and impurities. It also describes specific tests that may be included depending on the dosage form, such as dissolution, disintegration, hardness/friability for solid oral dosage forms. The guidelines provide information on justifying acceptance criteria and setting specifications based on development data and stability studies.
















































































