








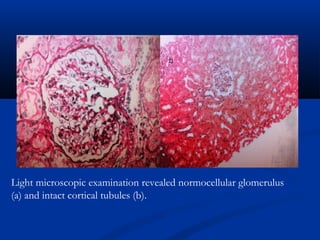

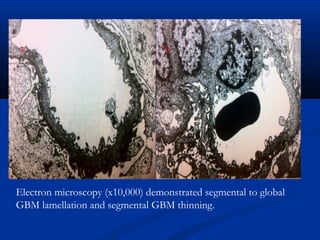











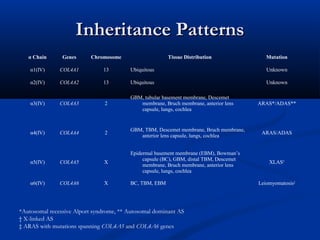









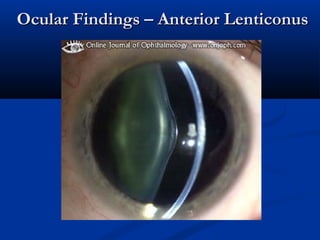




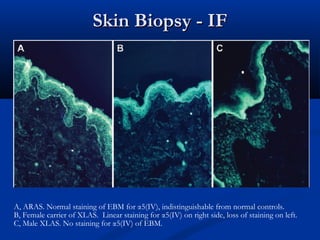
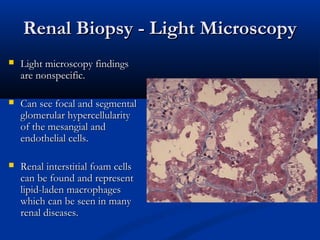


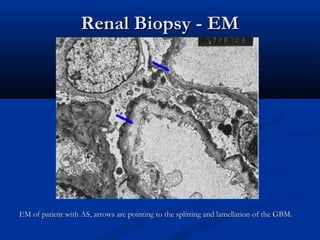
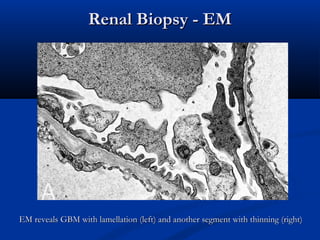
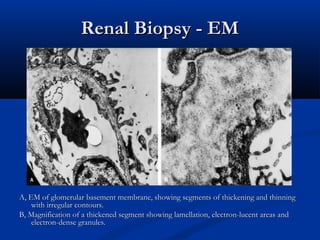







This document describes a case of a 19-year-old male who presented with proteinuria and microscopic hematuria. Renal biopsy revealed glomerulus and tubules appeared normal on light microscopy but electron microscopy showed glomerular basement membrane lamellation and thinning, consistent with a diagnosis of autosomal dominant Alport syndrome. The patient was prescribed lisinopril and showed stable kidney function on follow up. The background provided on Alport syndrome discusses its inheritance patterns, pathophysiology involving mutations in type IV collagen genes, and clinical features.






























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)








![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)



