

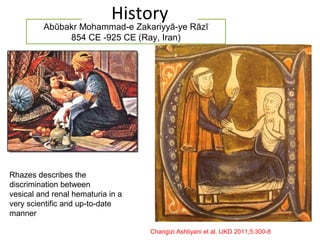


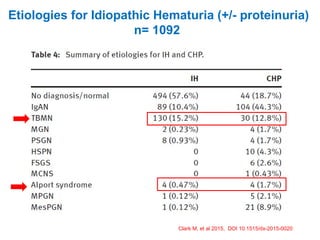


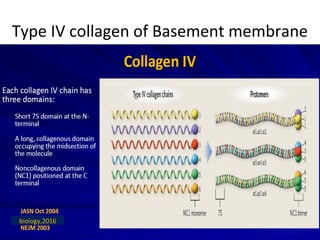
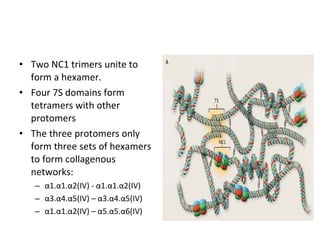

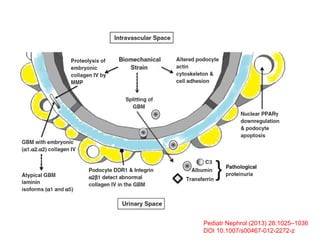
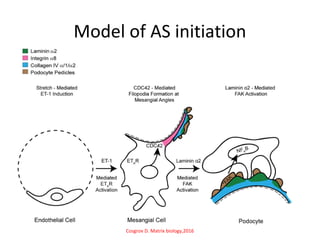
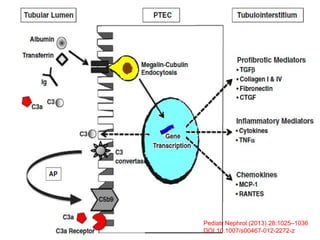

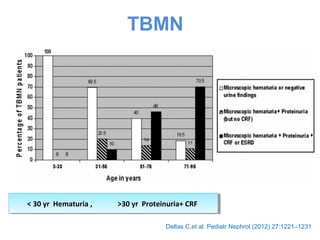

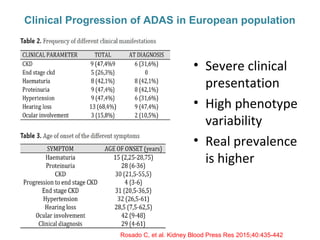

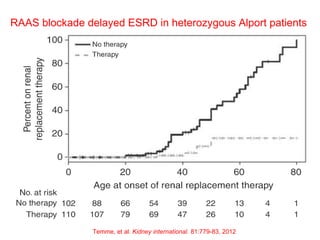
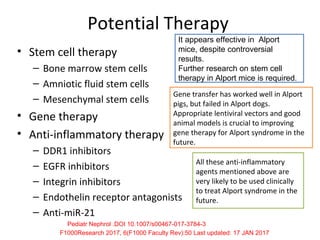
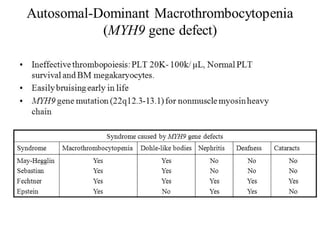

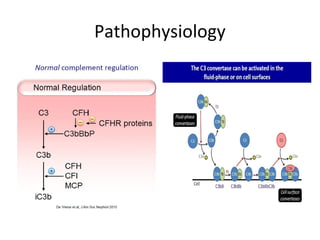

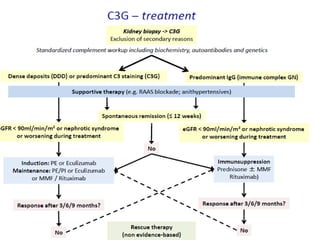



1. Familial hematuria refers to hereditary kidney diseases that cause microscopic or gross hematuria. Alport syndrome is a common cause and is a progressive hereditary glomerular basement membrane disease caused by mutations in collagen IV genes. 2. Angiotensin-converting enzyme inhibition is the first-line treatment for Alport syndrome to delay progression to end-stage renal disease, with angiotensin receptor blockers and aldosterone inhibition as second-line treatments. 3. Renal transplantation is curative for patients who progress to end-stage renal disease from Alport syndrome and results in good transplant outcomes. Long-term follow up is important given the risk of progression in familial































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)













![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)




