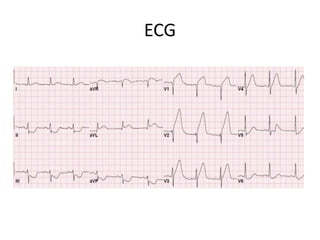
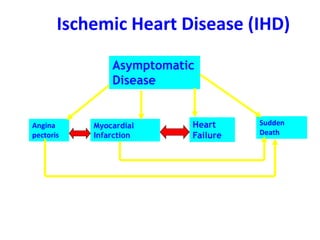
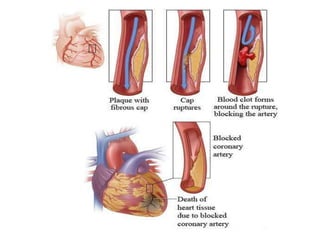
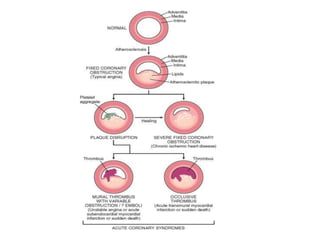
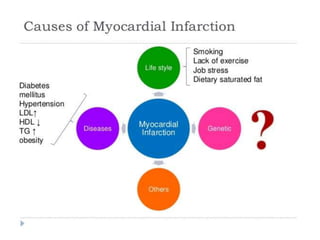
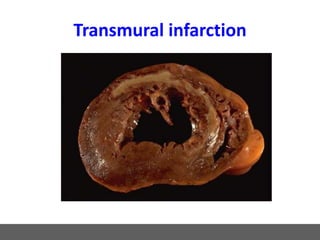
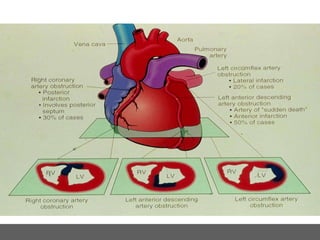
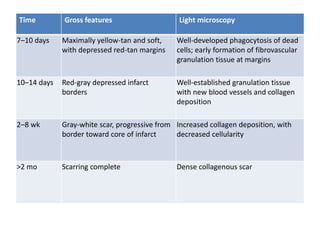
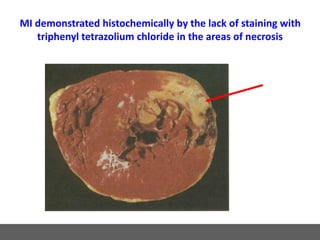
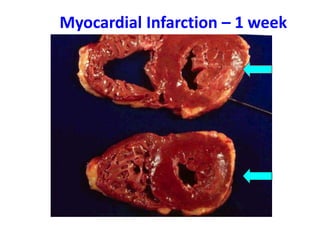
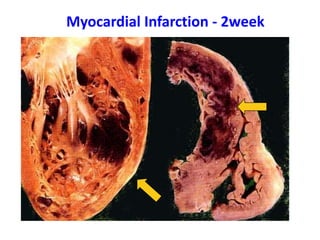
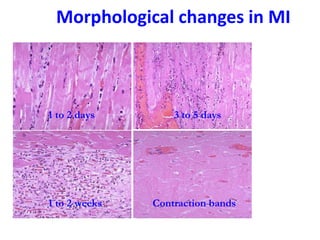
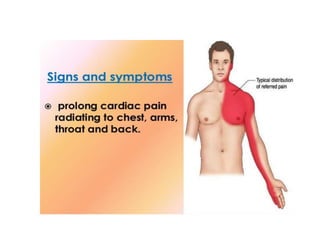
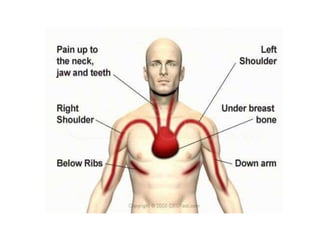
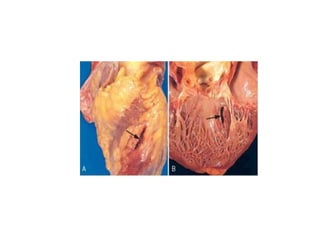
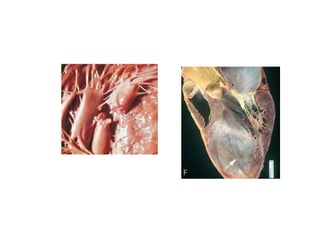
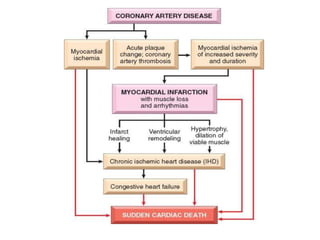
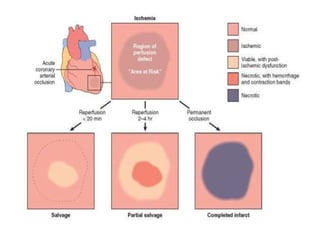
The patient presented with symptoms of chest pain, shortness of breath, and sweating. ECG and blood tests showed elevated cardiac markers. This is consistent with a diagnosis of myocardial infarction (MI). MI occurs when blood flow to the heart is blocked, causing heart muscle cell death. On pathology, MI presents as areas of necrosis and inflammation. Laboratory tests for MI diagnosis include cardiac troponins, CK-MB, and myoglobin, which are more specific and sensitive than total CK or LDH. Together, the presentation and test results make the diagnosis of MI likely in this case.