The document provides information on ischemic heart disease (IHD), including:
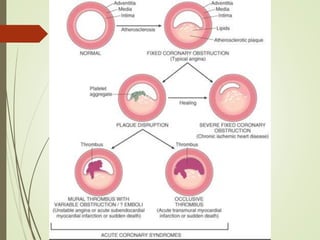
1) IHD is caused by inadequate blood supply to the heart muscle and can be due to blockages in the coronary arteries from atherosclerosis or other causes like vasospasm.
2) IHD can present as stable angina, unstable angina, myocardial infarction, or heart failure. A myocardial infarction occurs when prolonged ischemia causes death of heart muscle tissue.
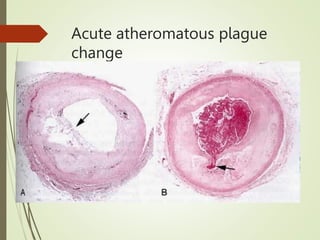
3) The pathology of a myocardial infarction involves plaque rupture, thrombus formation, and complete blockage of blood flow leading to irreversible damage to heart muscle within minutes to hours.