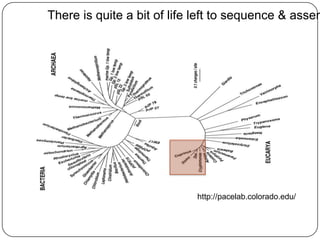
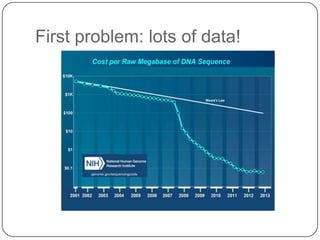
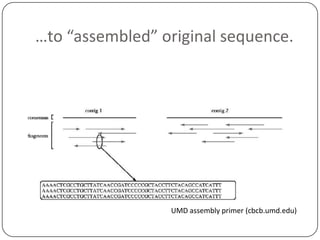
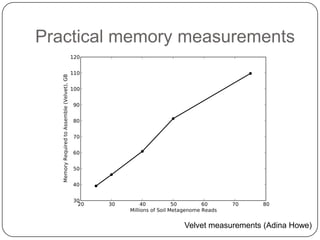
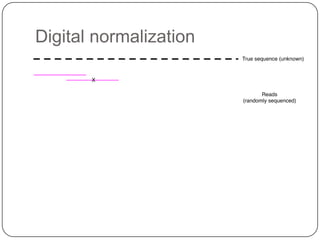
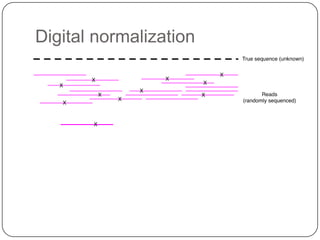
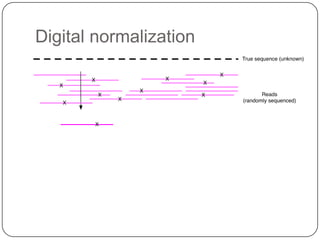
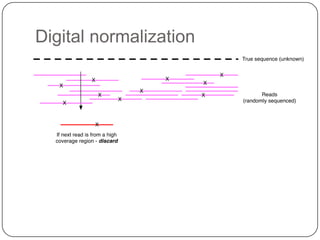
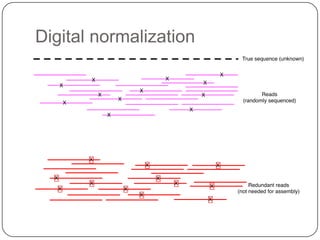
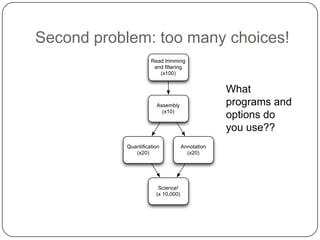
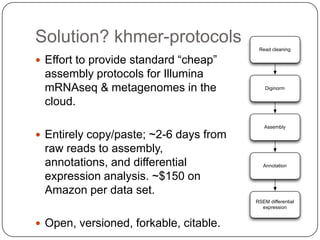
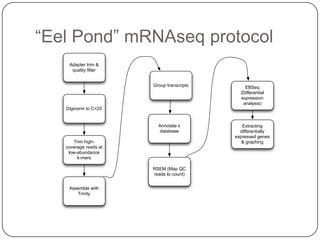
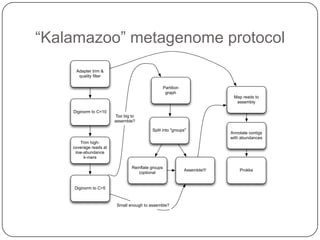
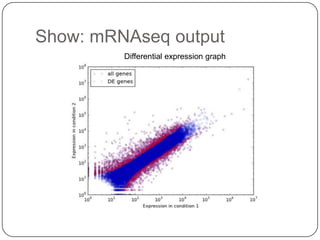
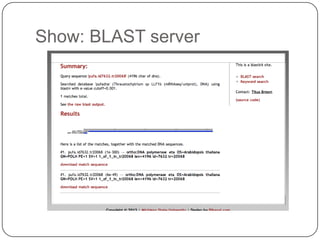
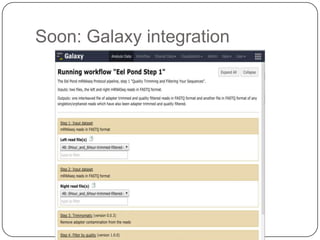
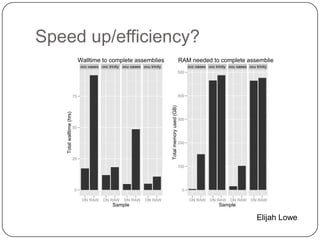
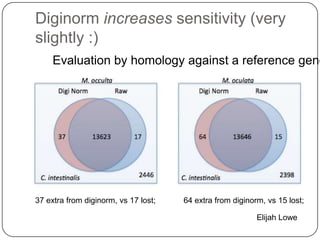
The document discusses three main problems with de novo assembly of next generation sequencing data and proposes solutions. The three problems are 1) large memory and compute requirements for assembly, 2) complexity of the assembly process and lack of standardized protocols, and 3) limited training opportunities that are difficult for students. The proposed solutions are standardized assembly protocols called khmer-protocols that provide copy-paste workflows for mRNAseq and metagenome assembly using techniques like digital normalization to reduce memory usage and make assembly scalable. The khmer-protocols are designed to be open, versioned, and reproducible to generate initial assembly results cheaply and easily in the cloud.