Downloaded 3,190 times



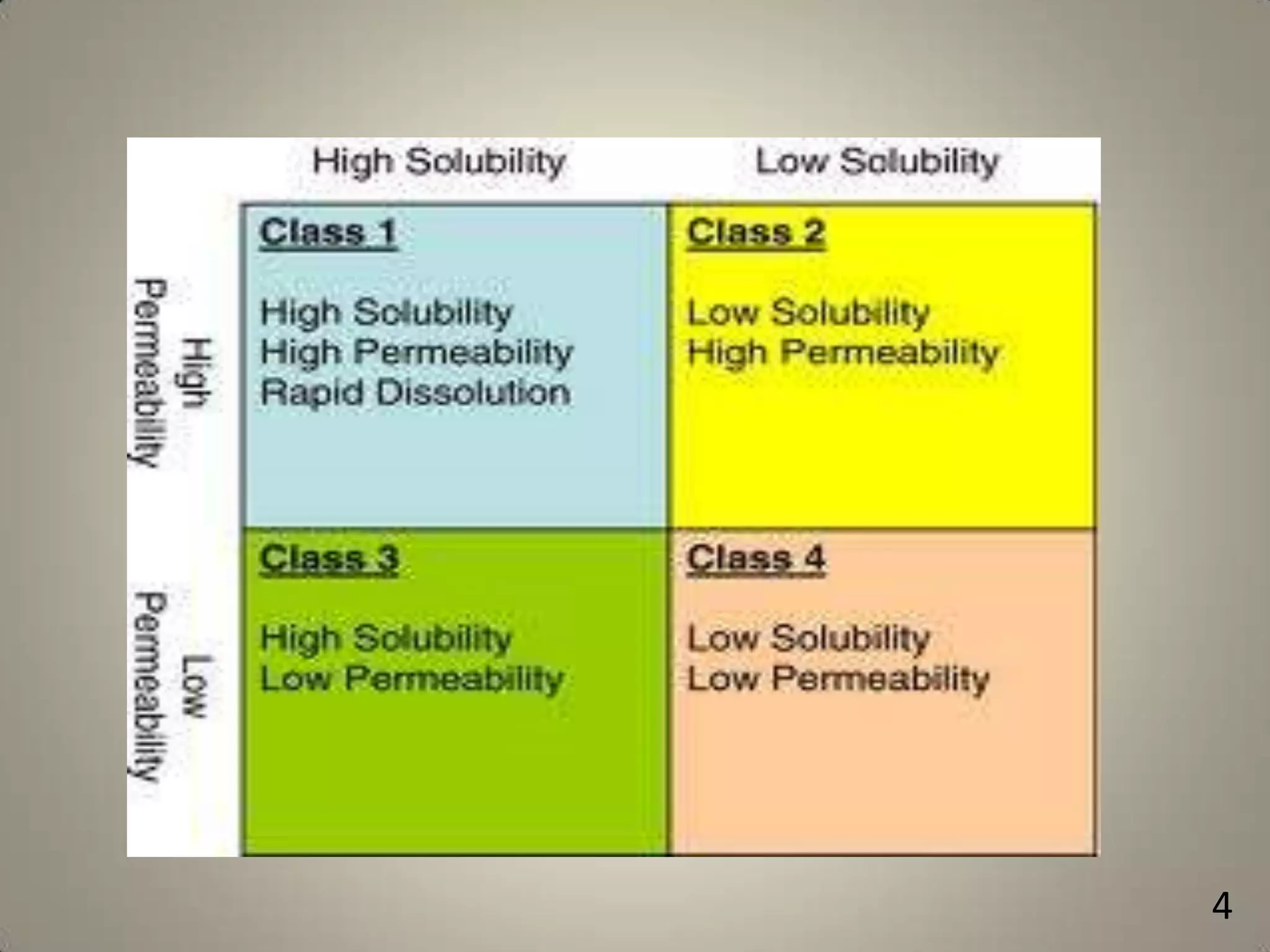
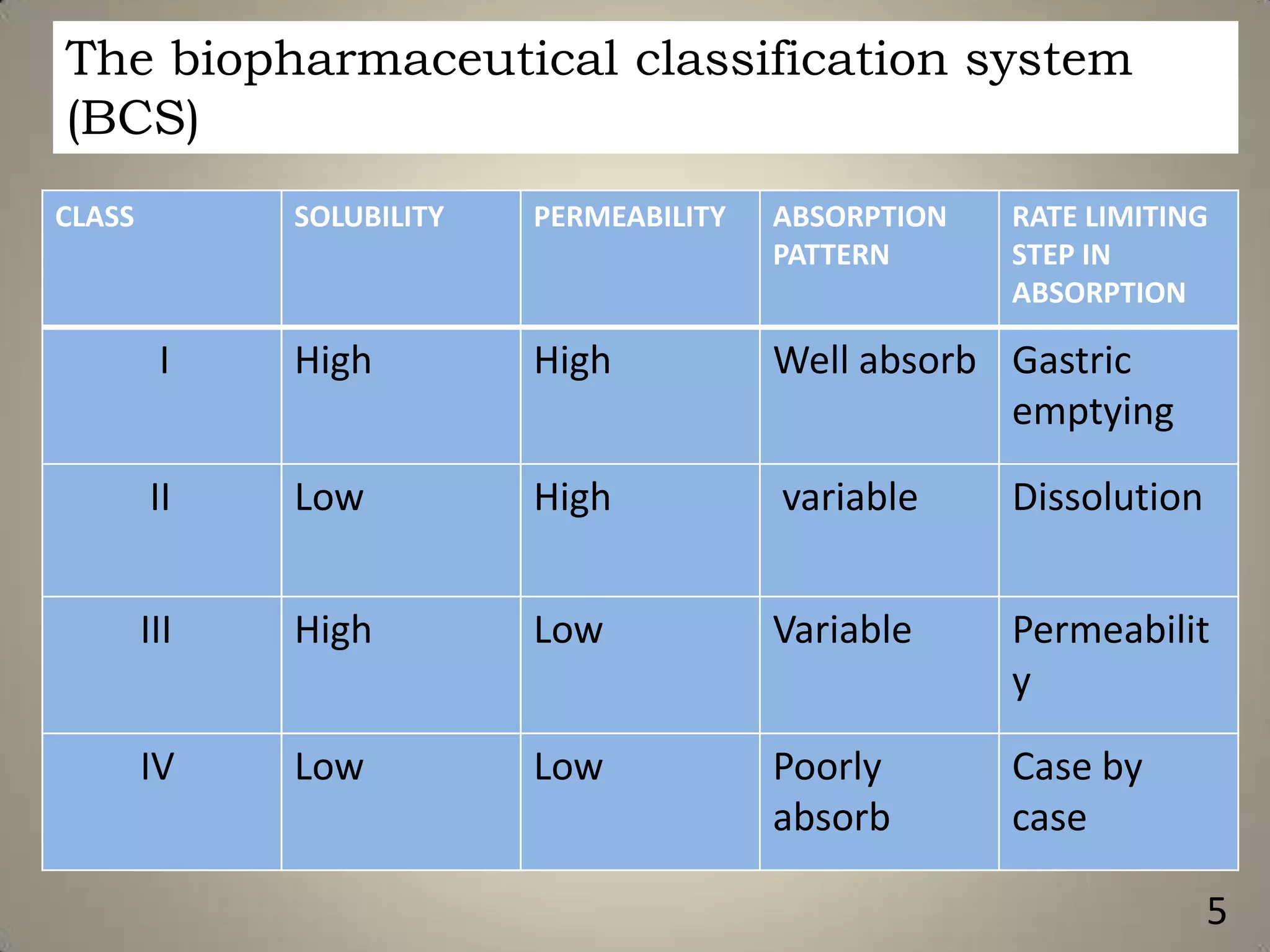
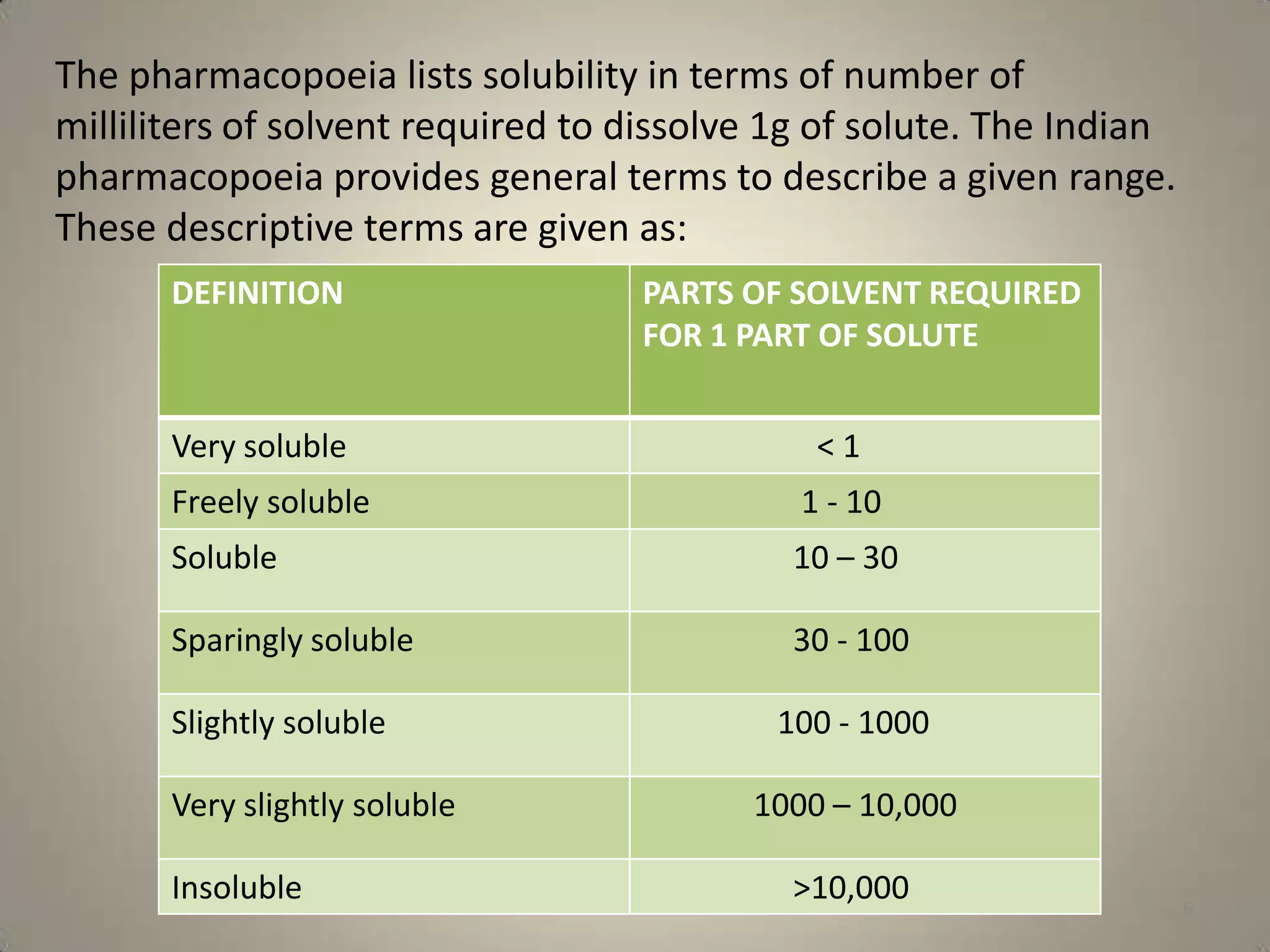

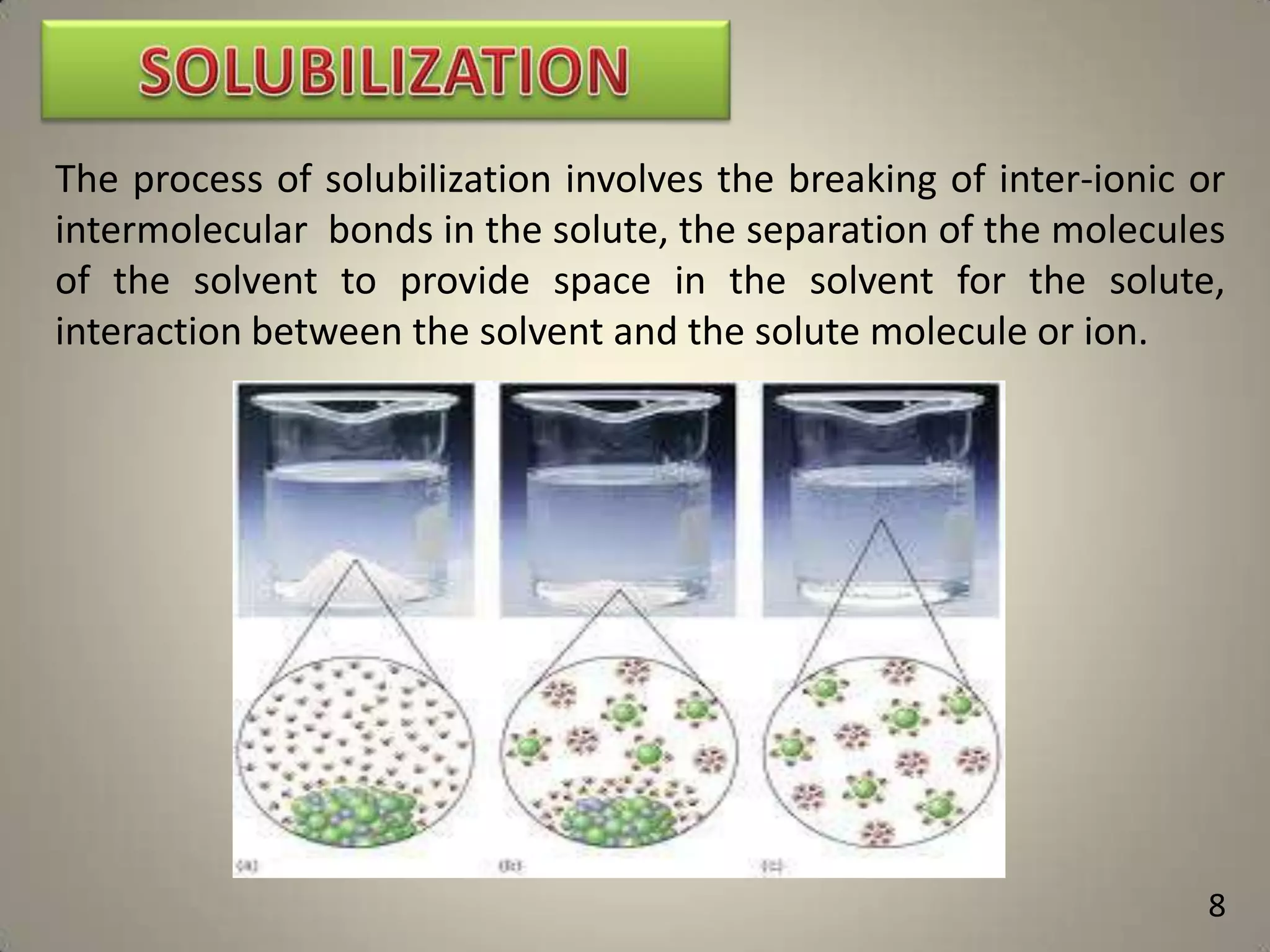
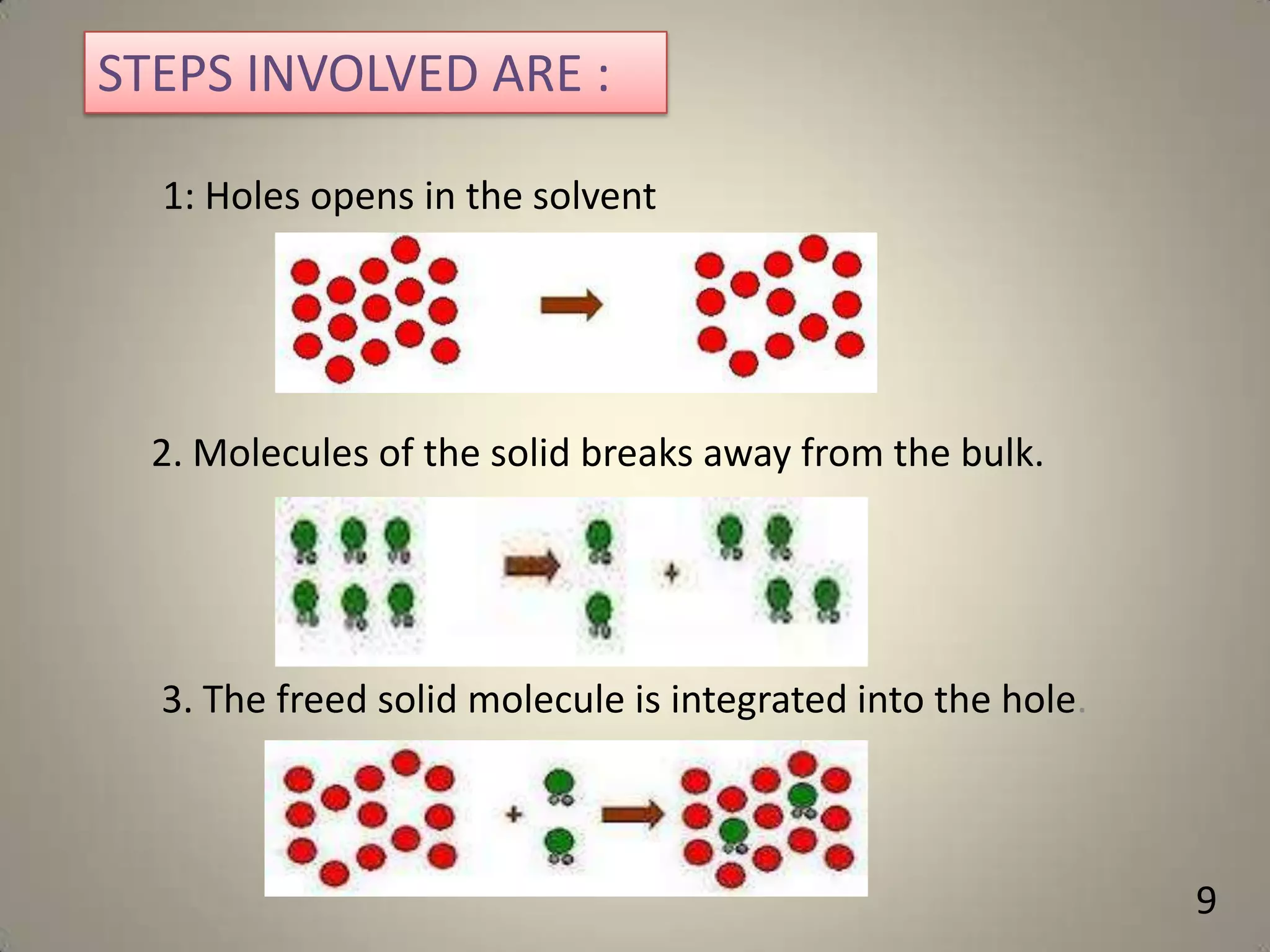





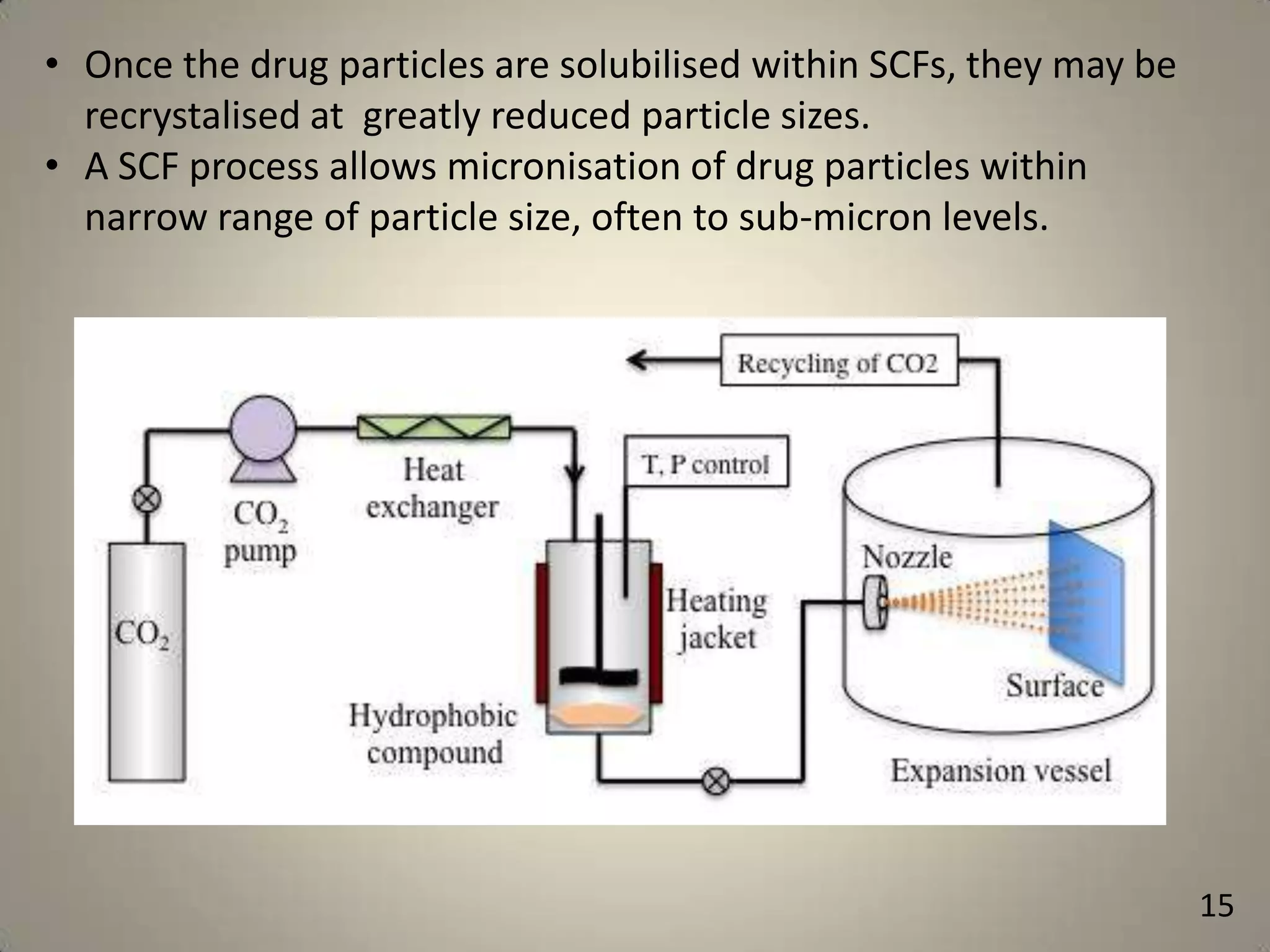



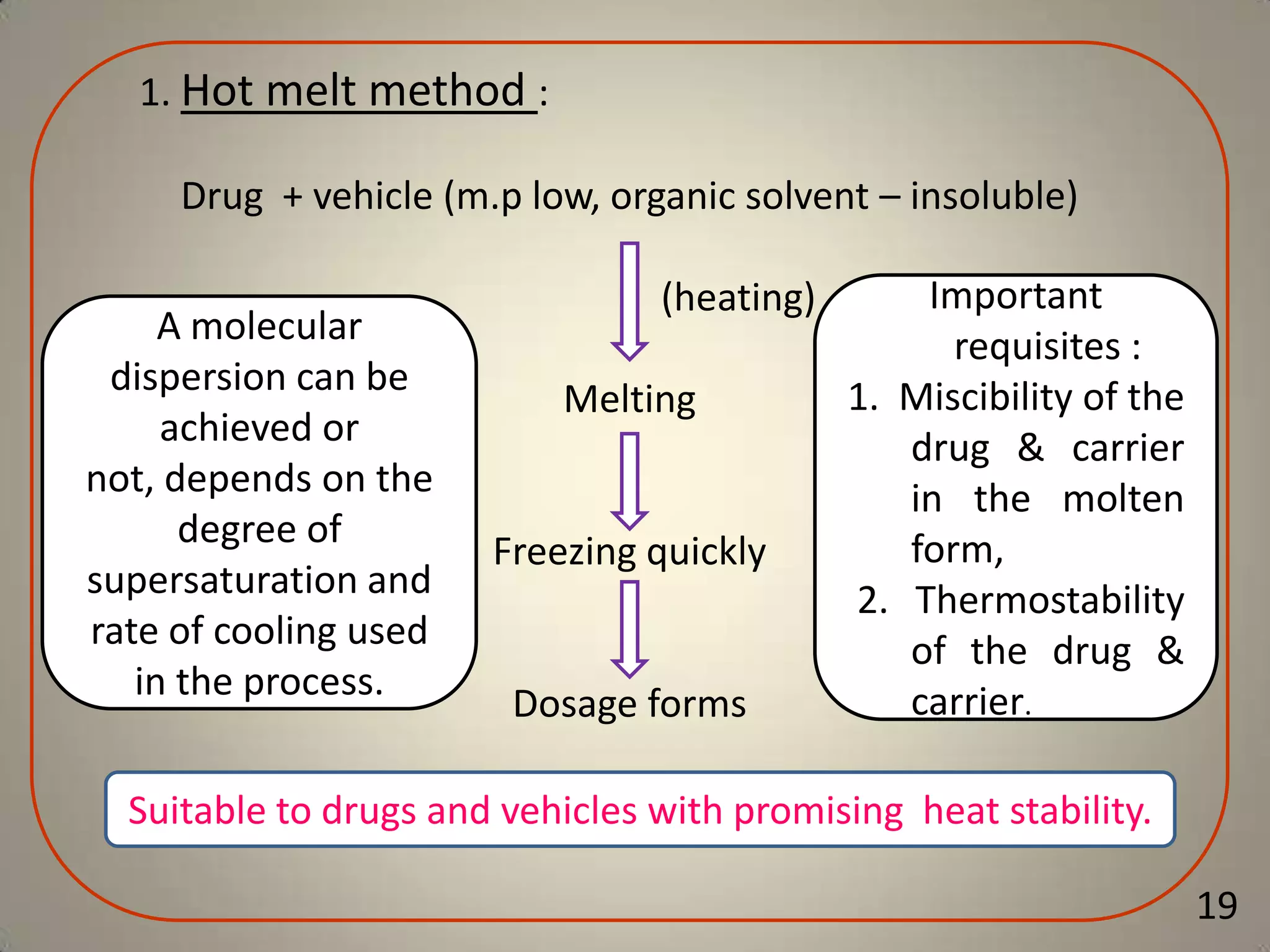


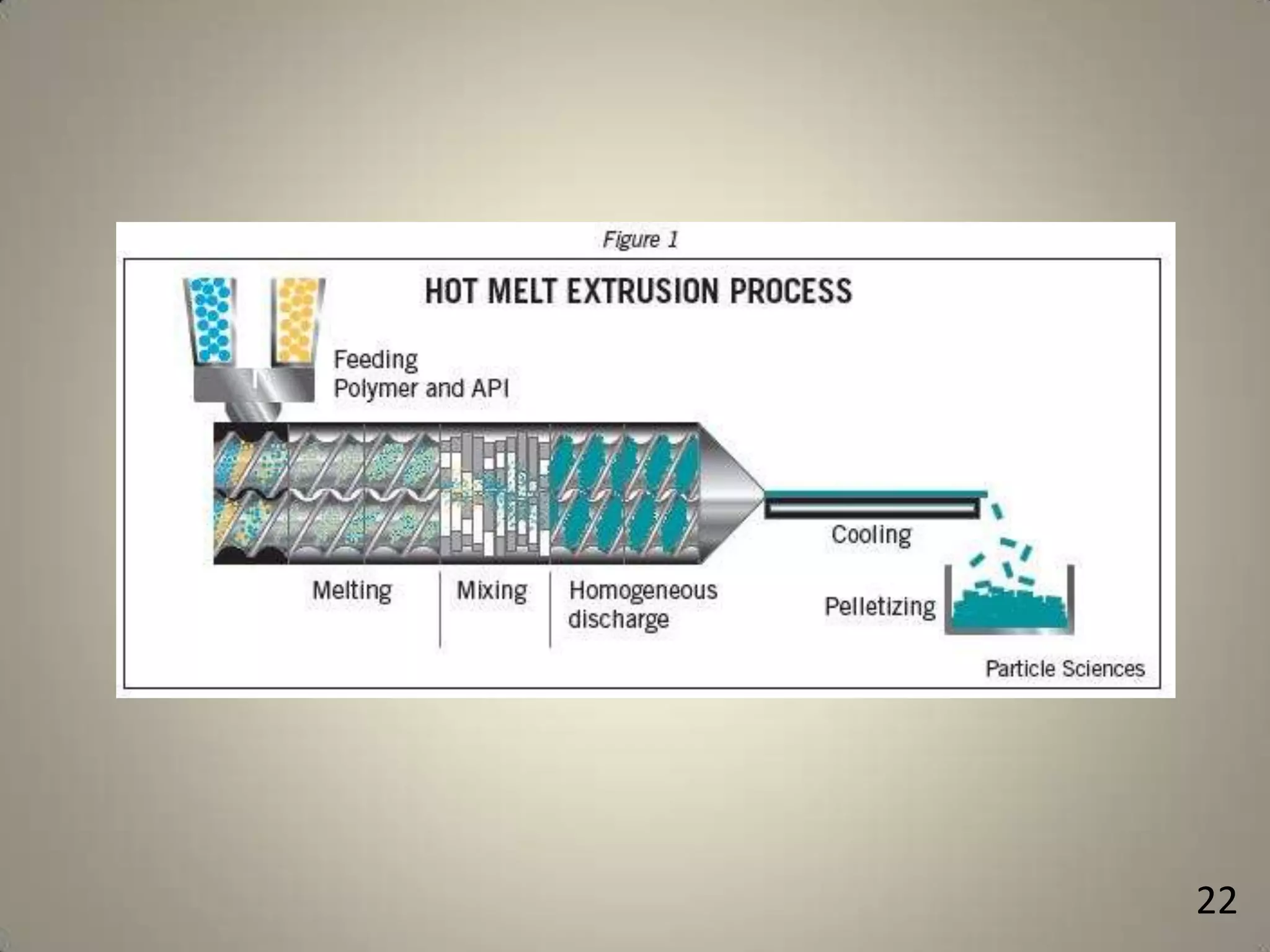
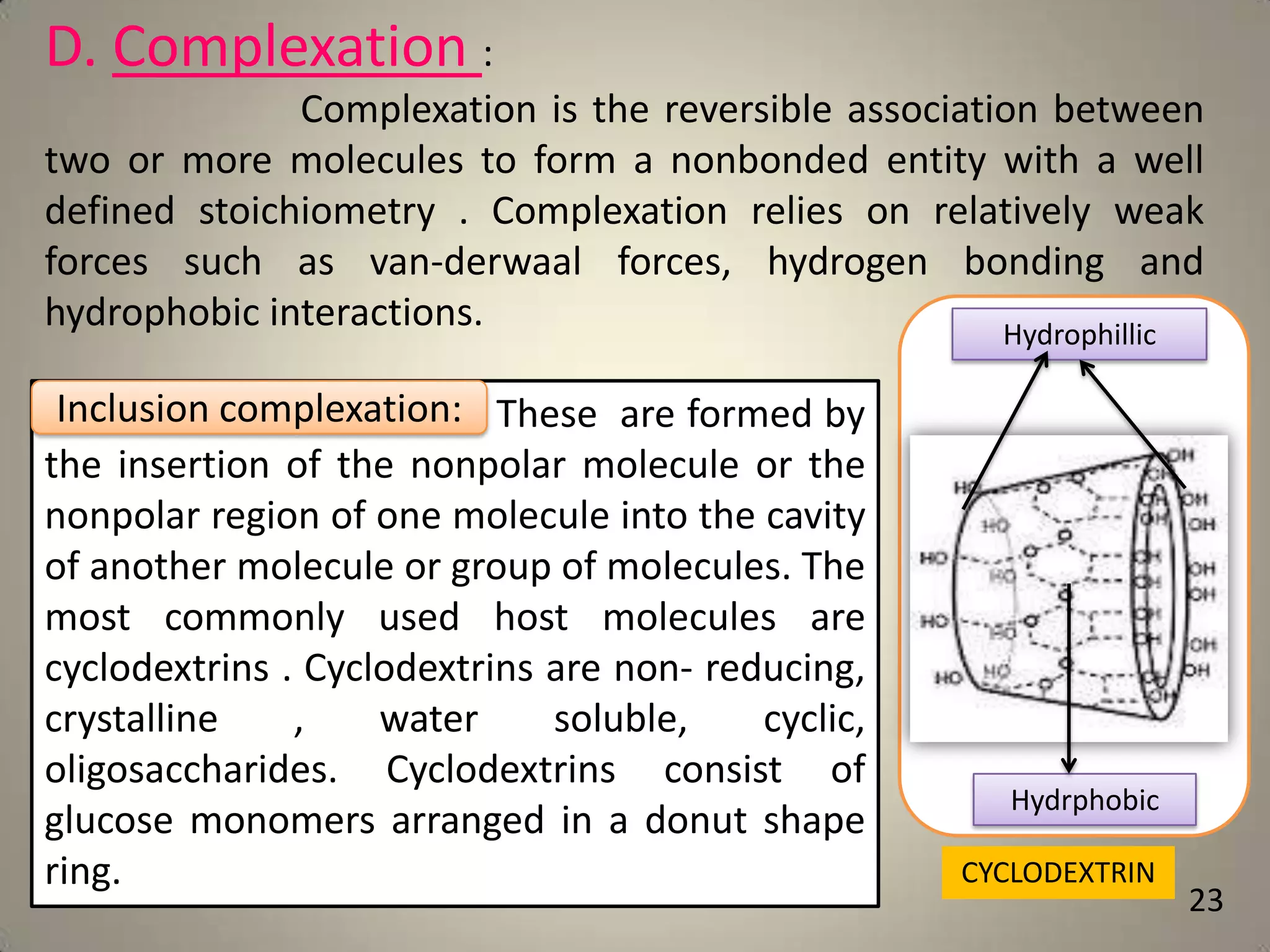

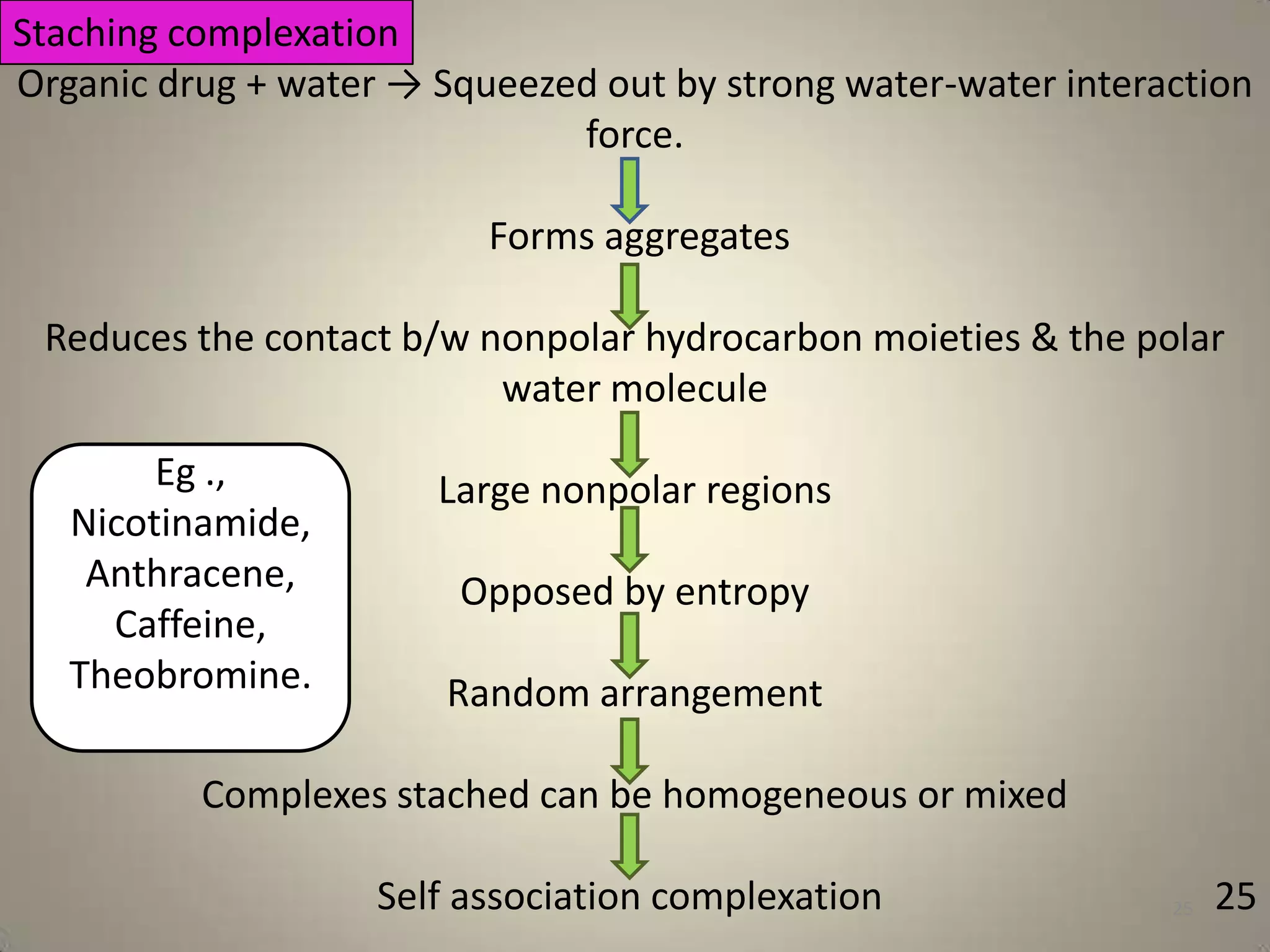
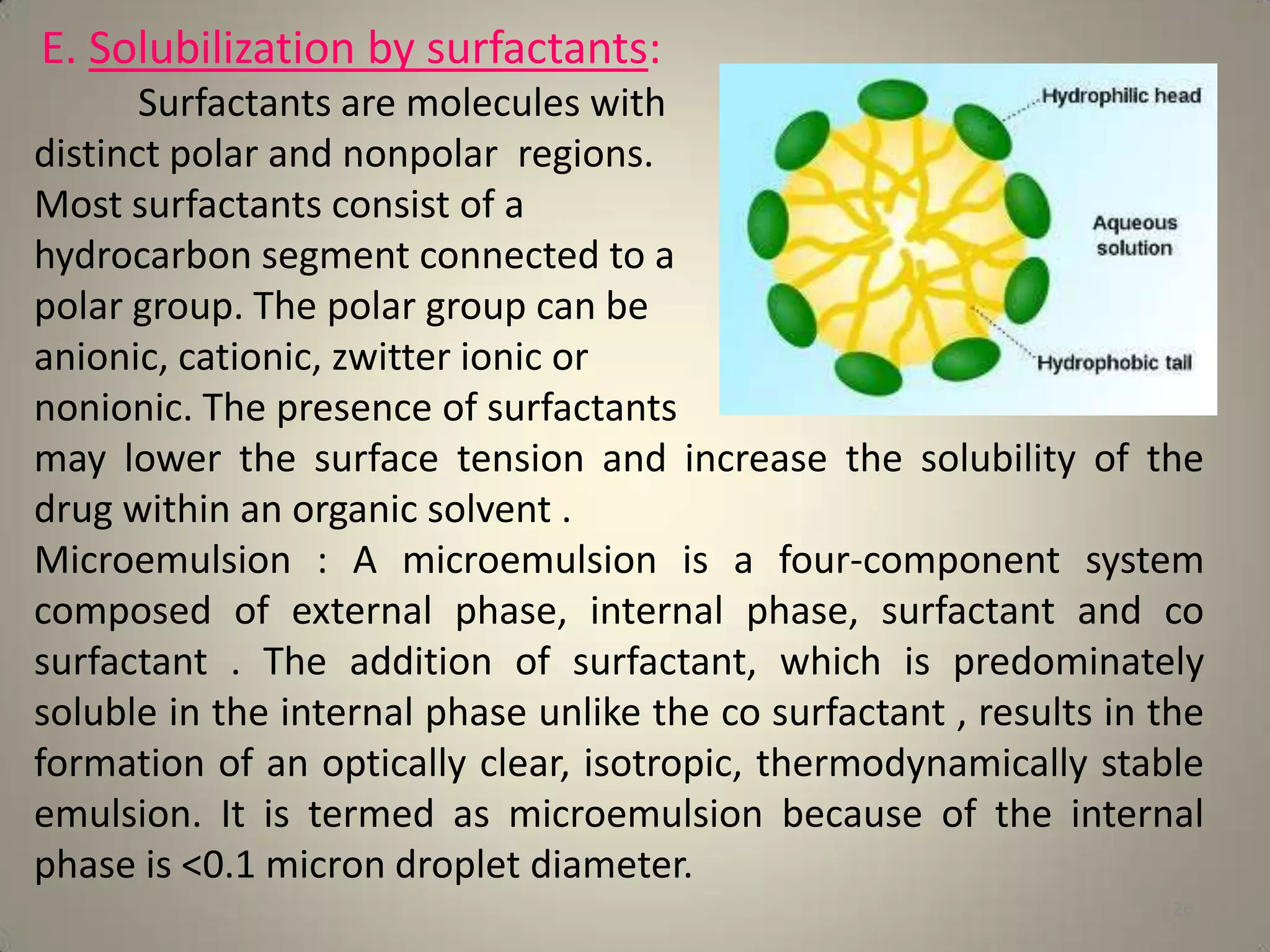





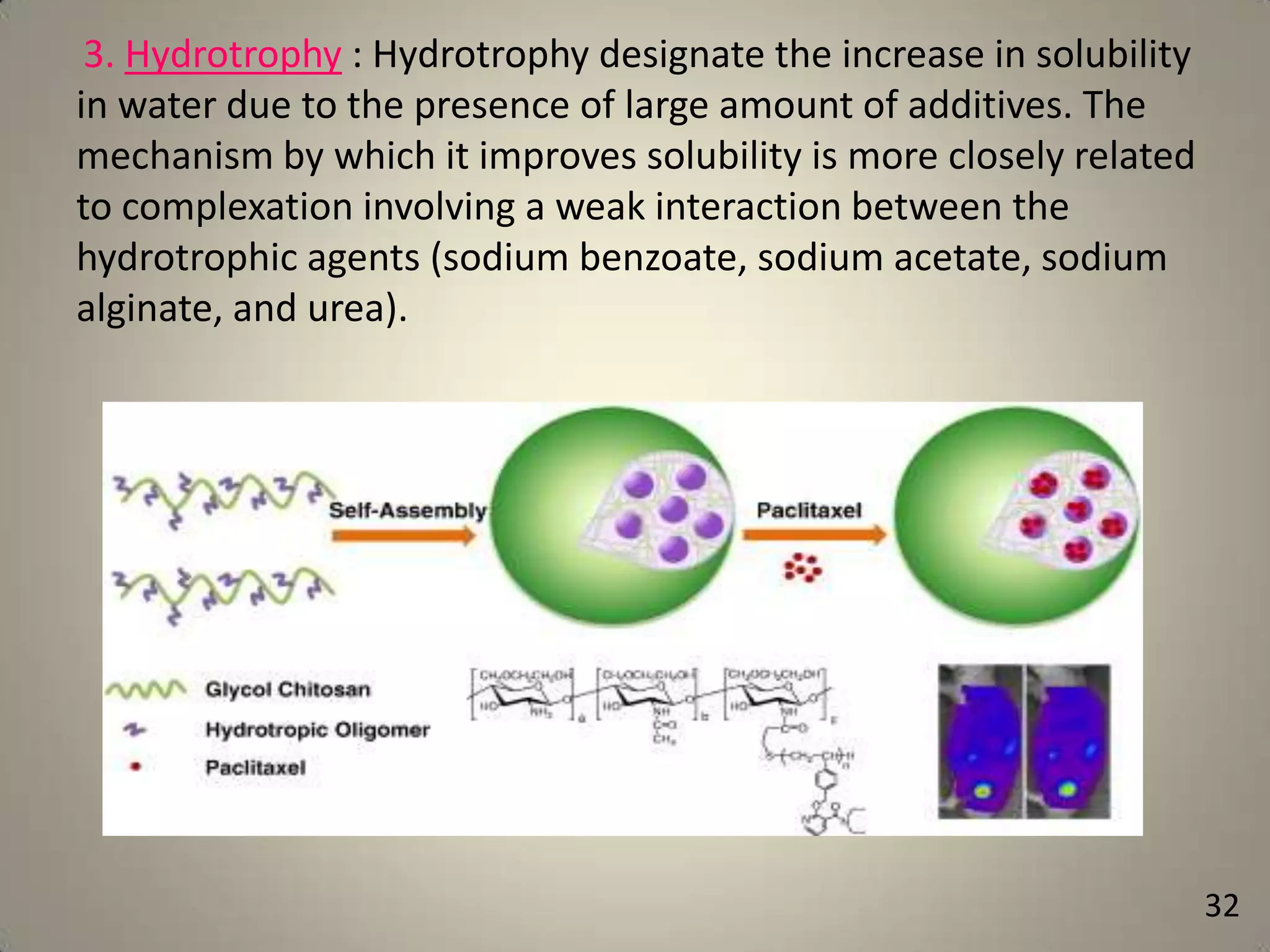














The document discusses various methods to improve drug solubility including physical modifications like particle size reduction through micronization or formation of nanosuspensions, modification of crystal habit through polymorphism, and drug dispersion in carriers through techniques like solid dispersions. It also discusses chemical modifications such as changing pH, use of buffers, and derivatization. Other methods covered are complexation, solubilization by surfactants to form microemulsions, co-crystallization, cosolvency, hydrotrophy, and solvent deposition. The biopharmaceutical classification system relating solubility and permeability to drug absorption is also summarized.

















































































