Download as PDF, PPTX
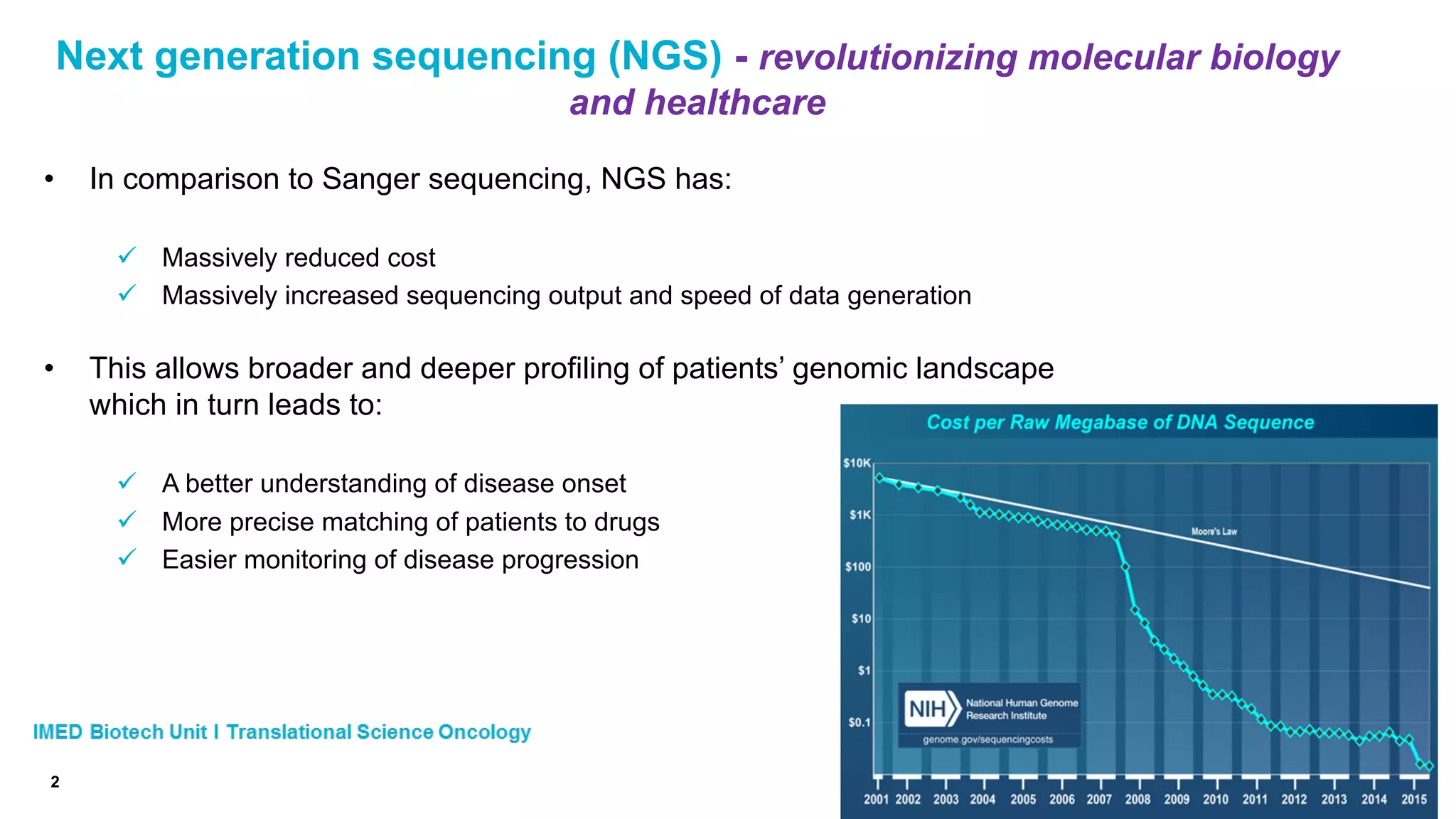
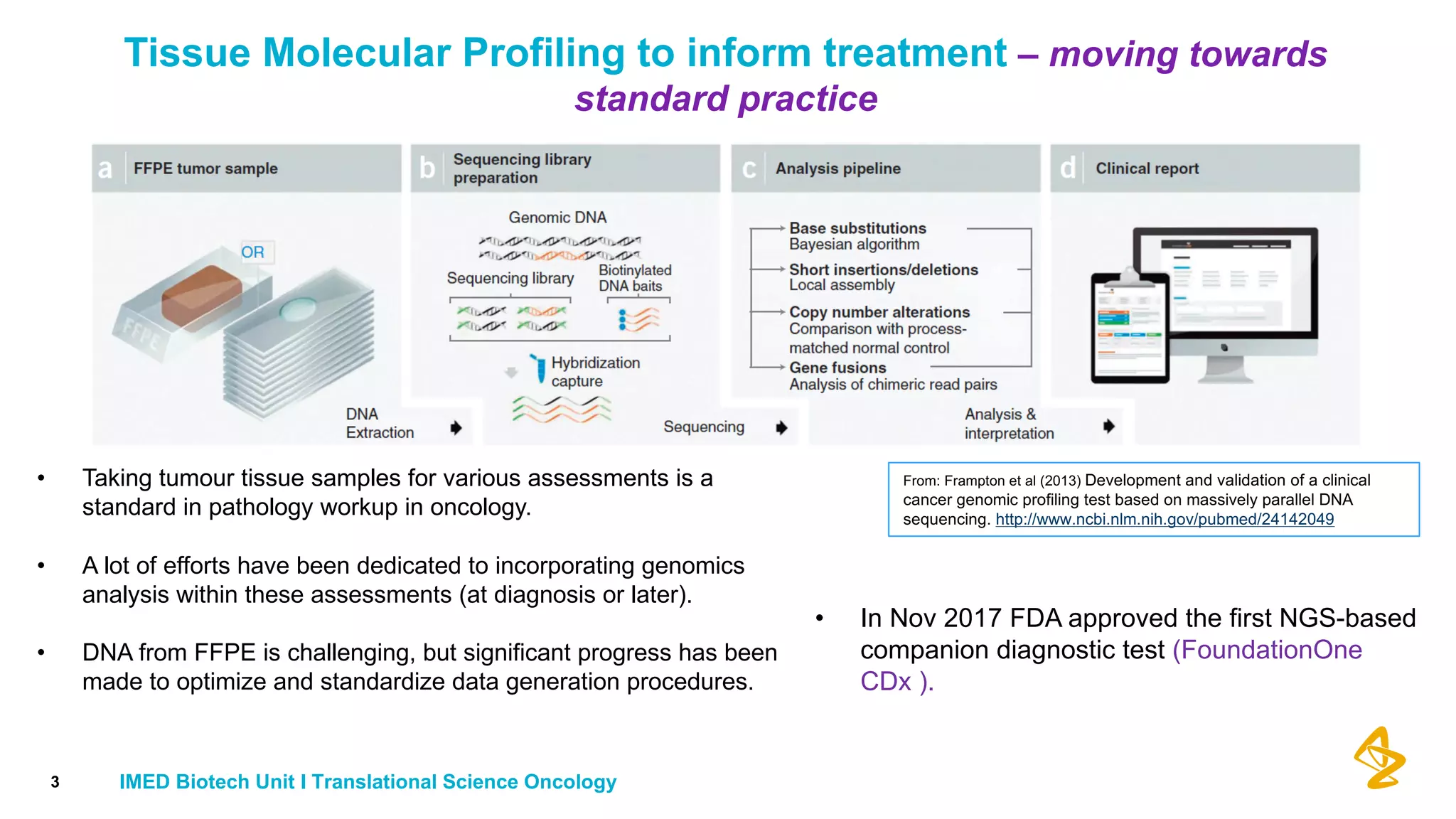
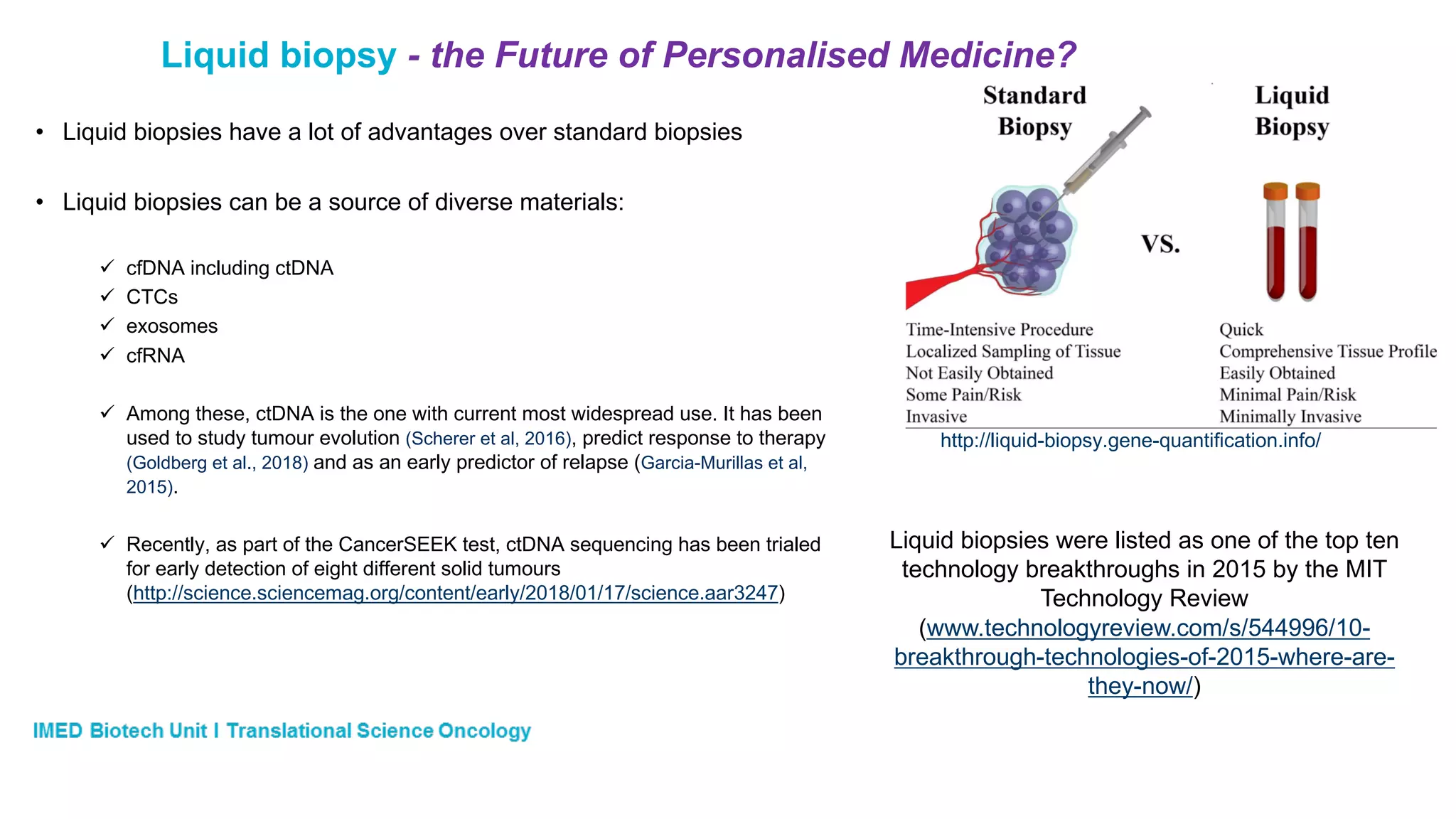

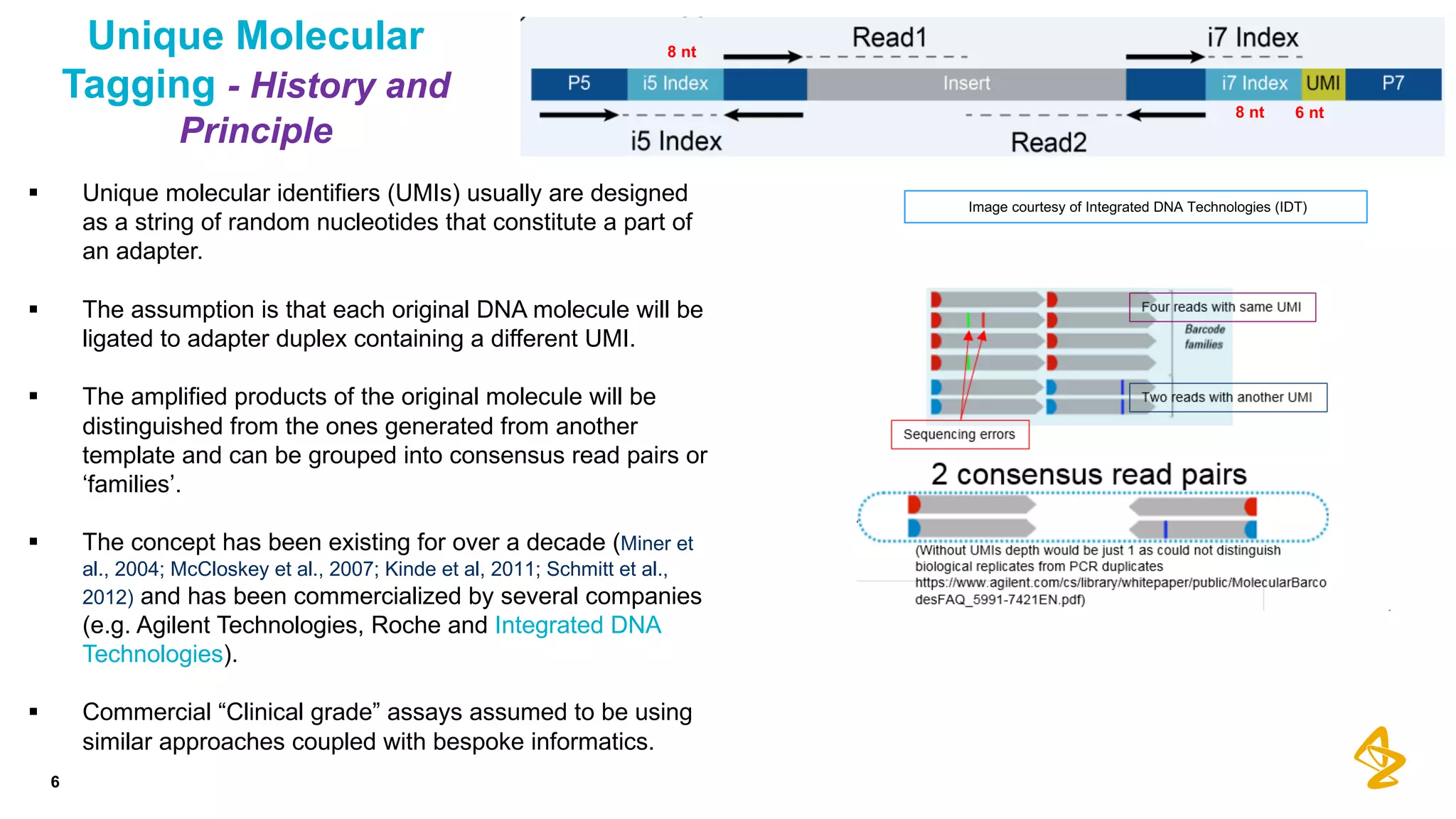

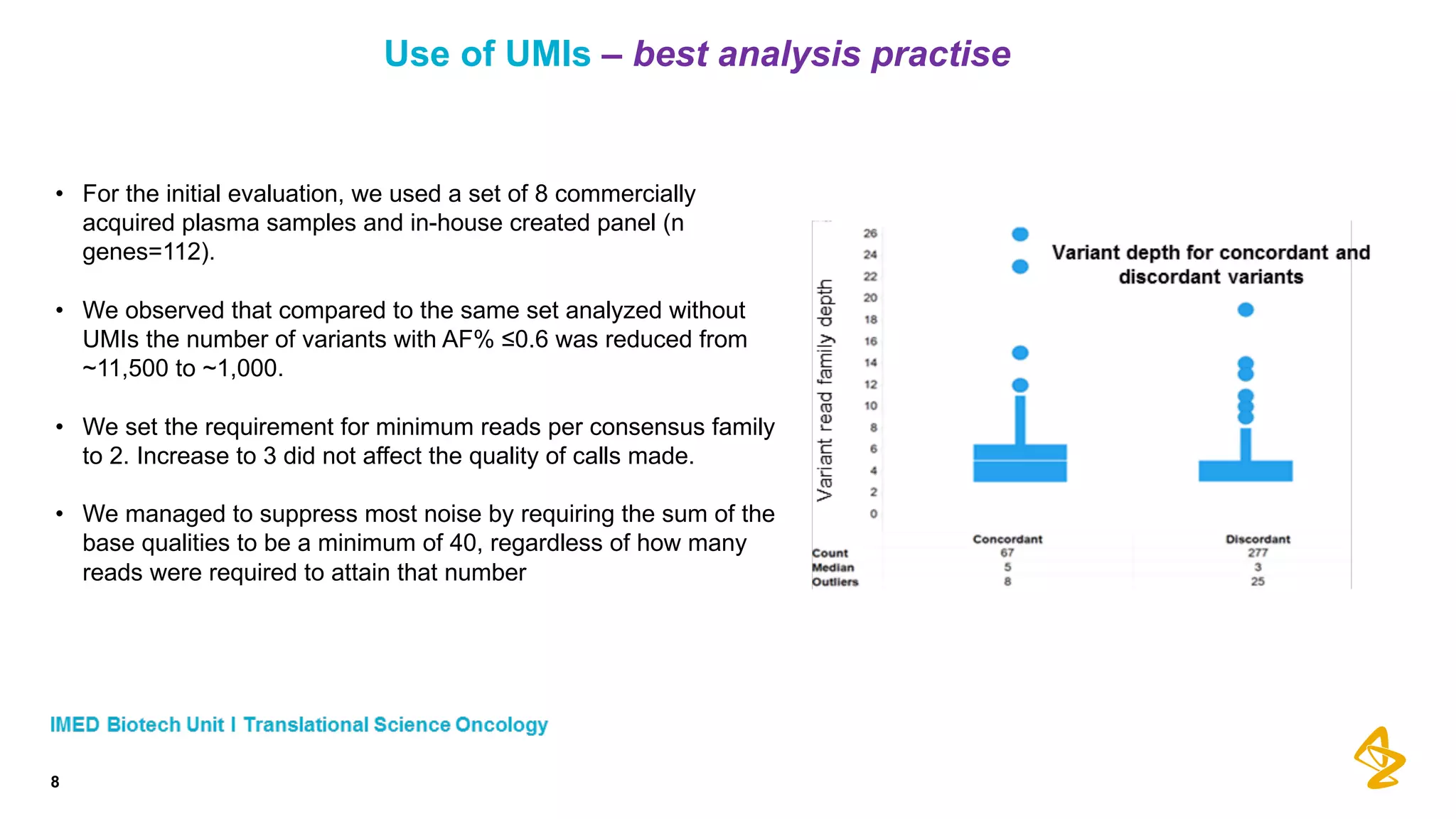
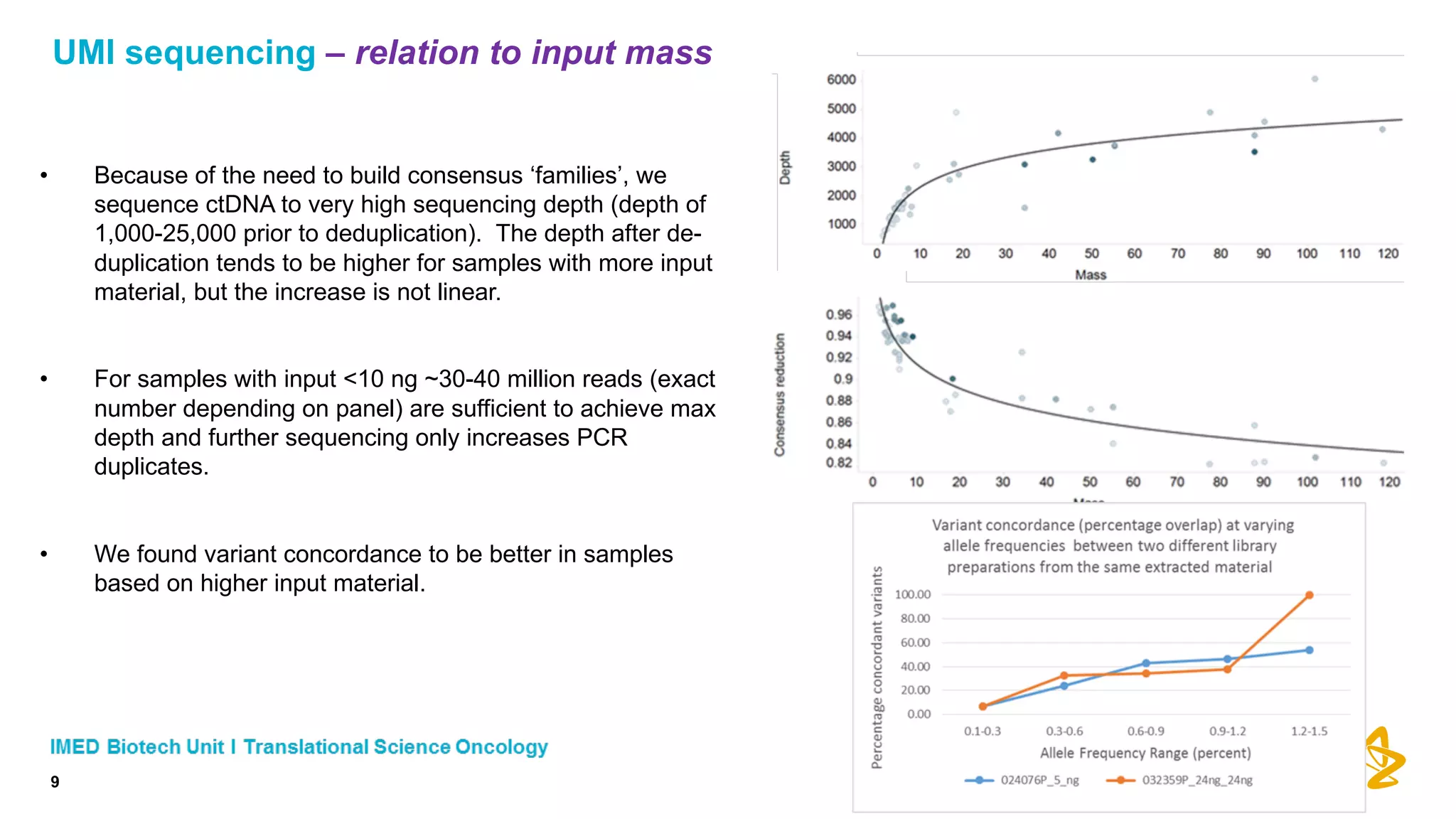
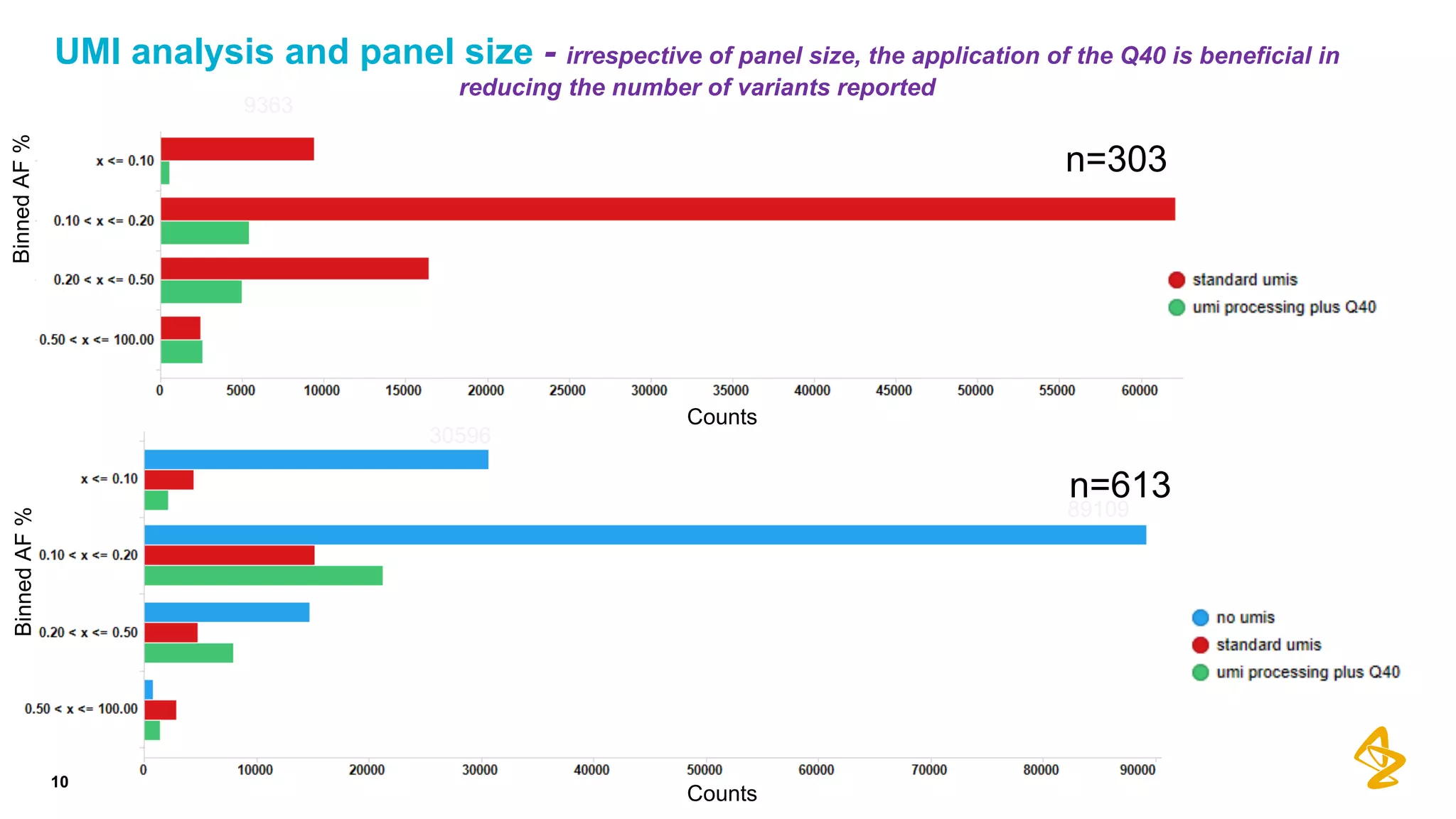
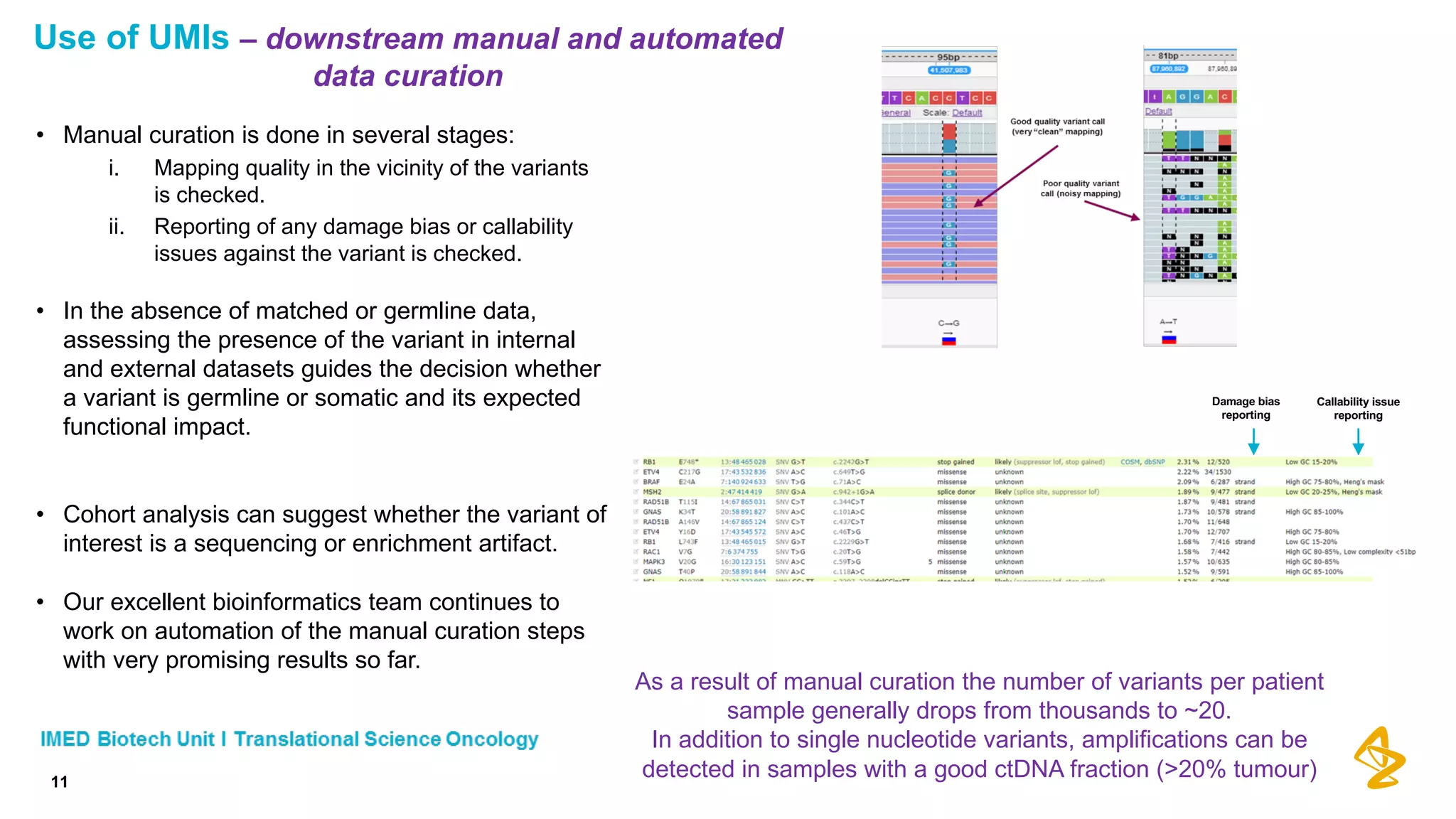
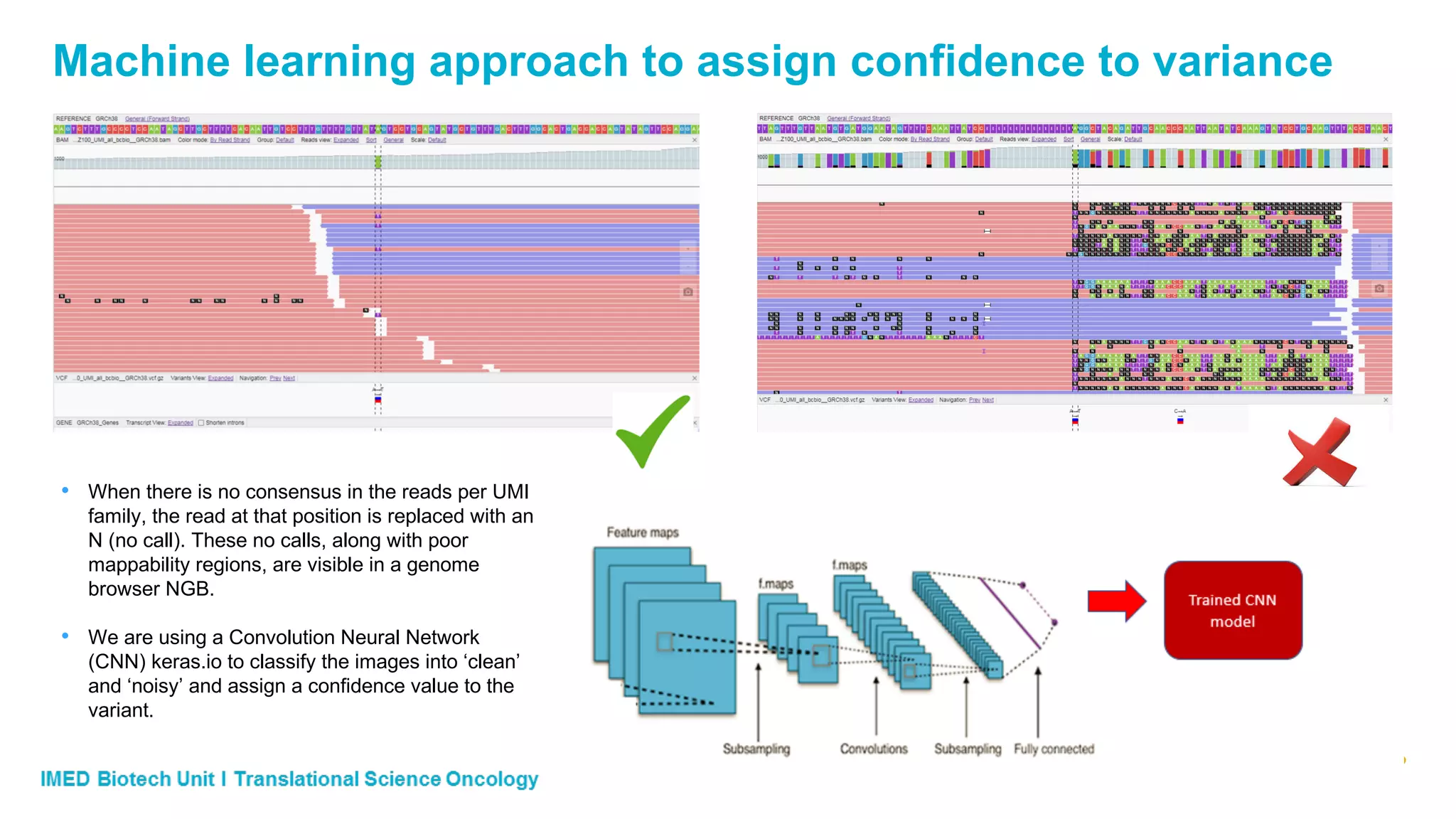
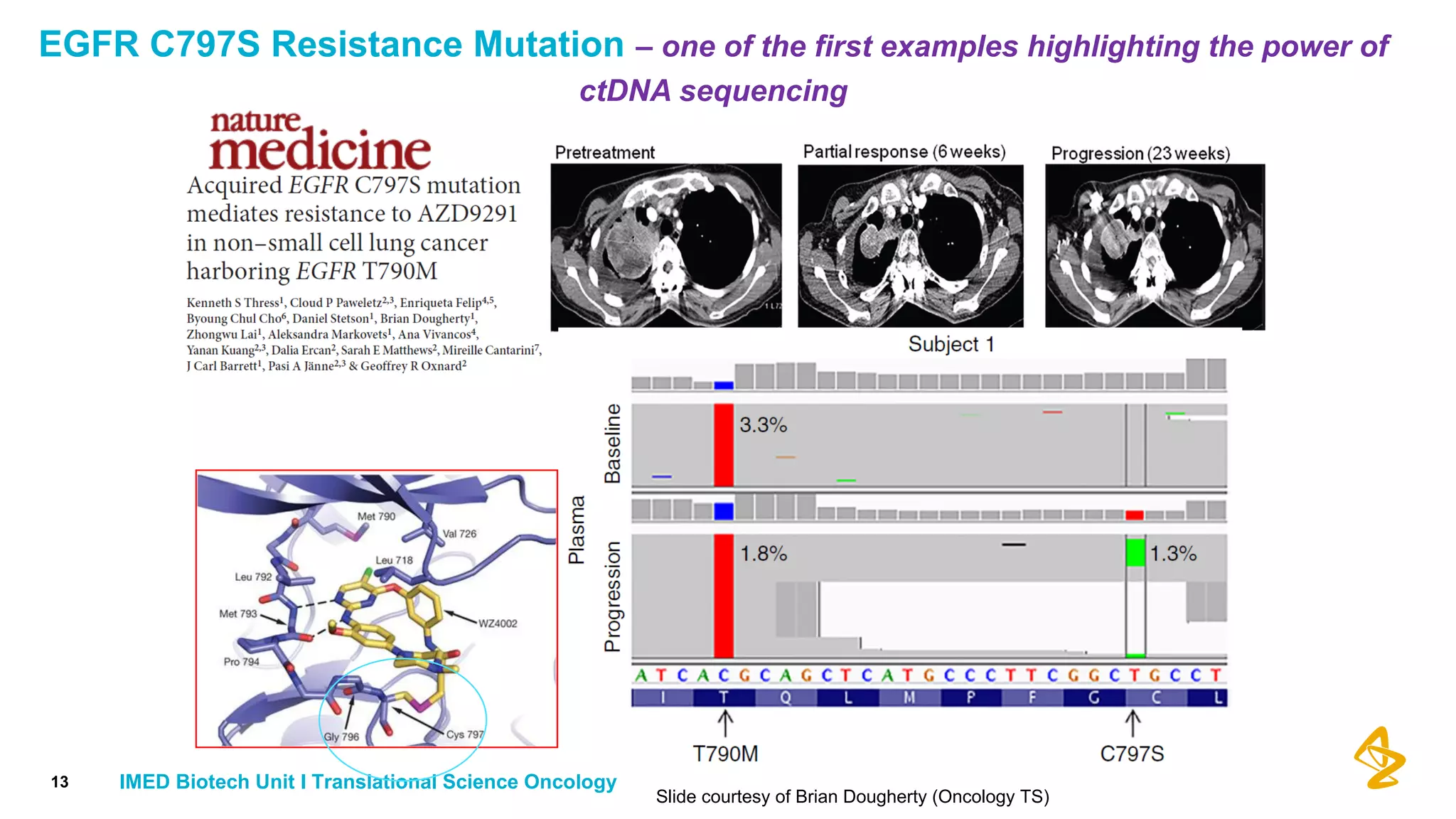
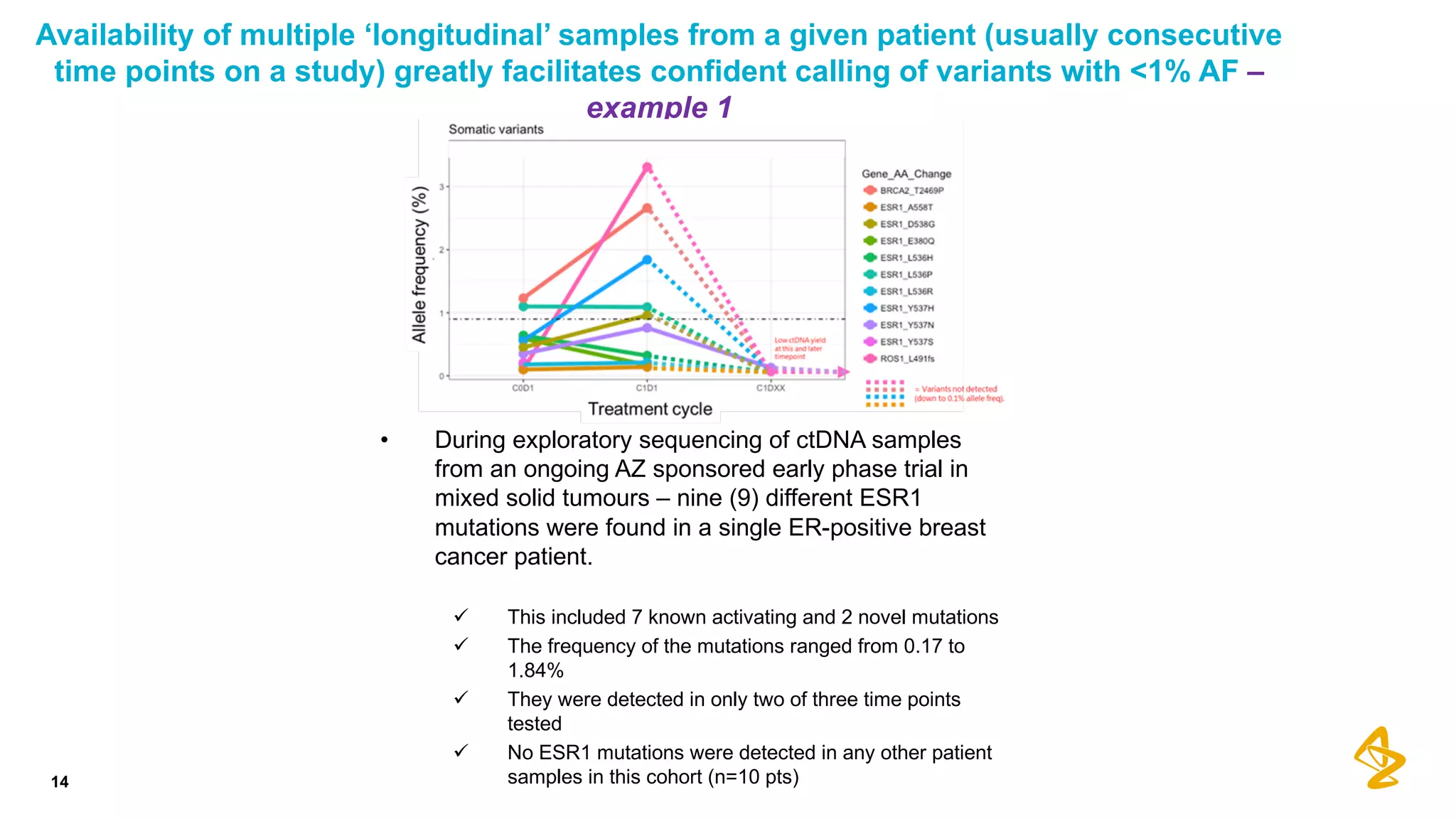
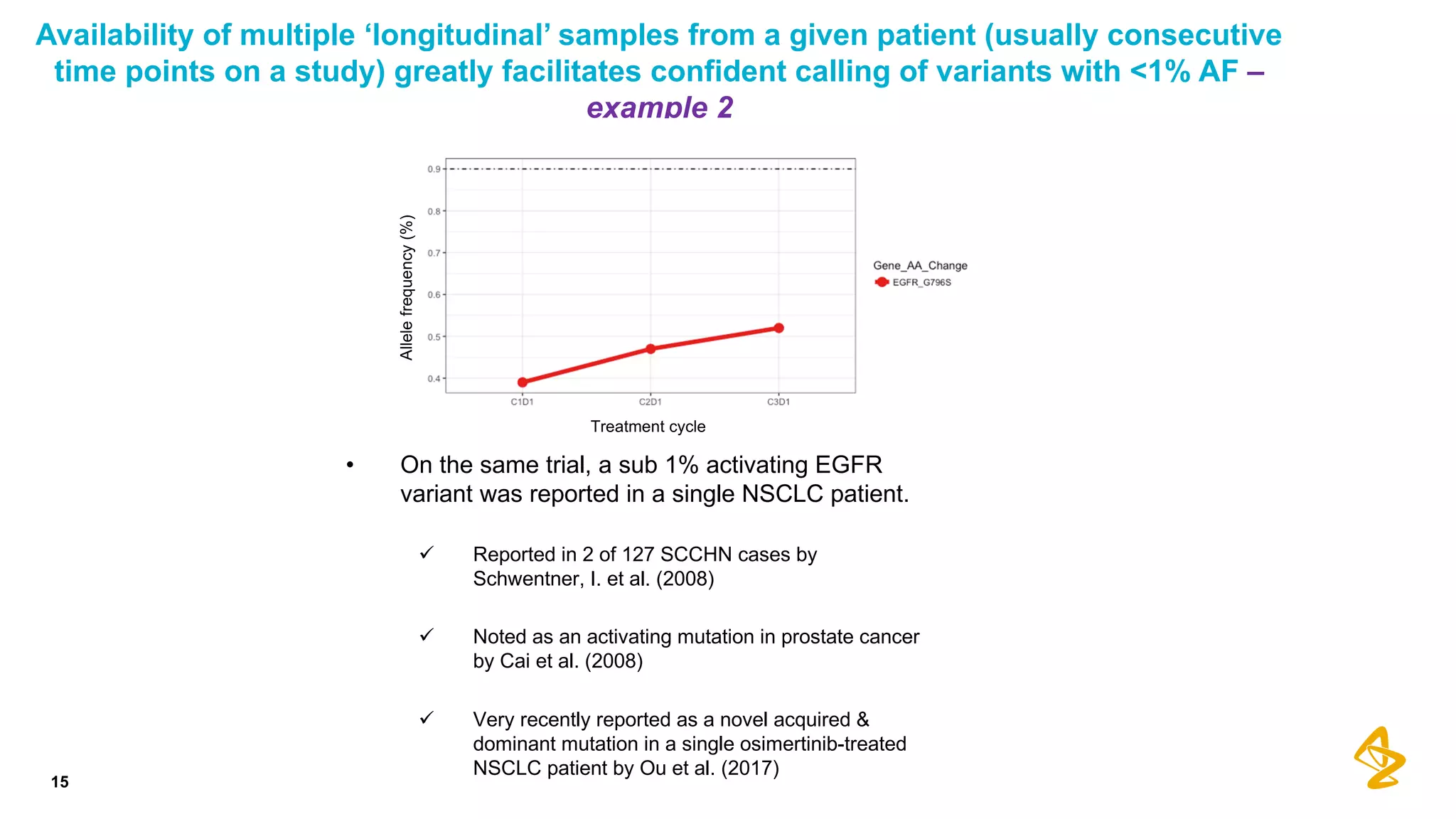
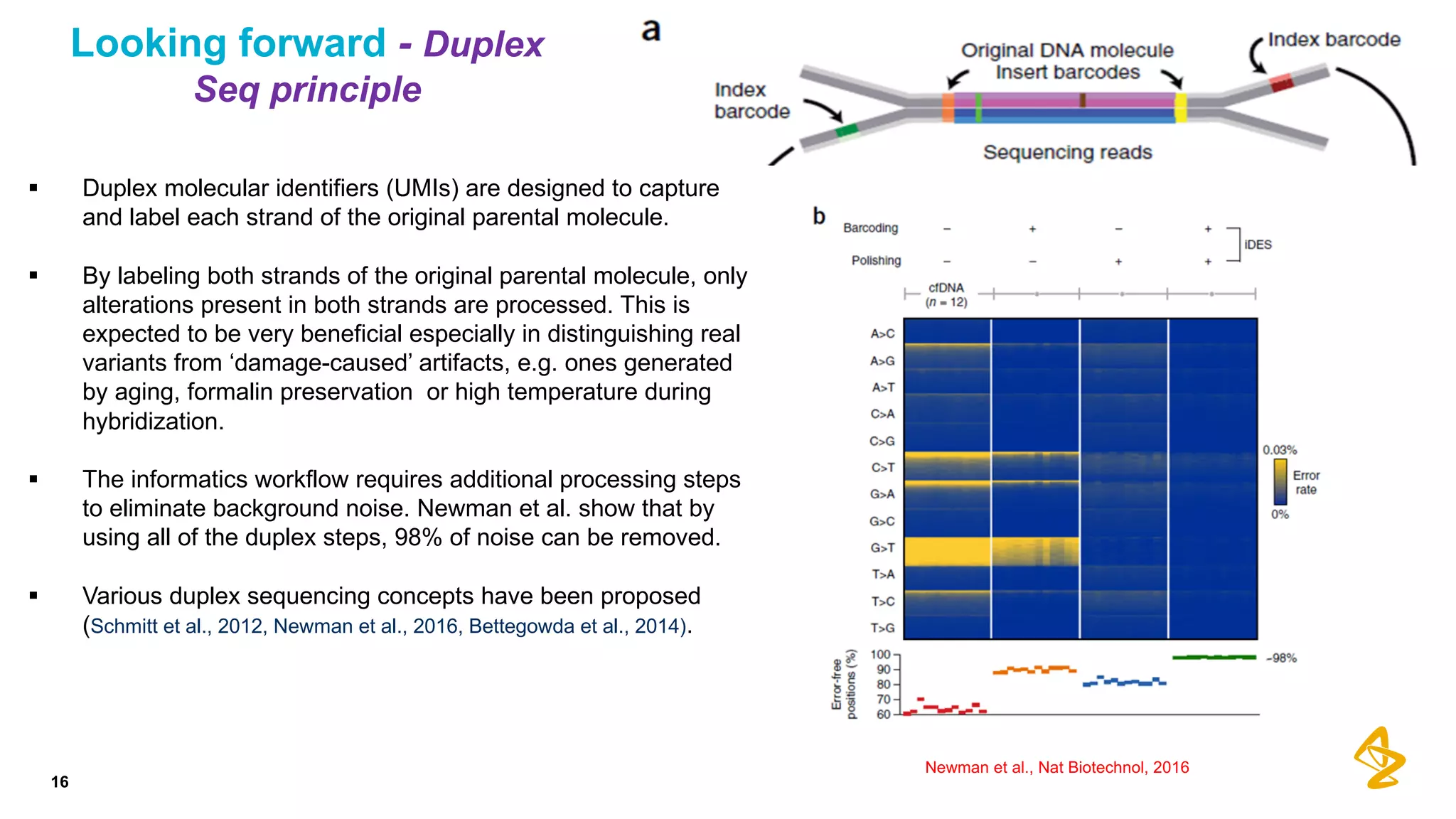
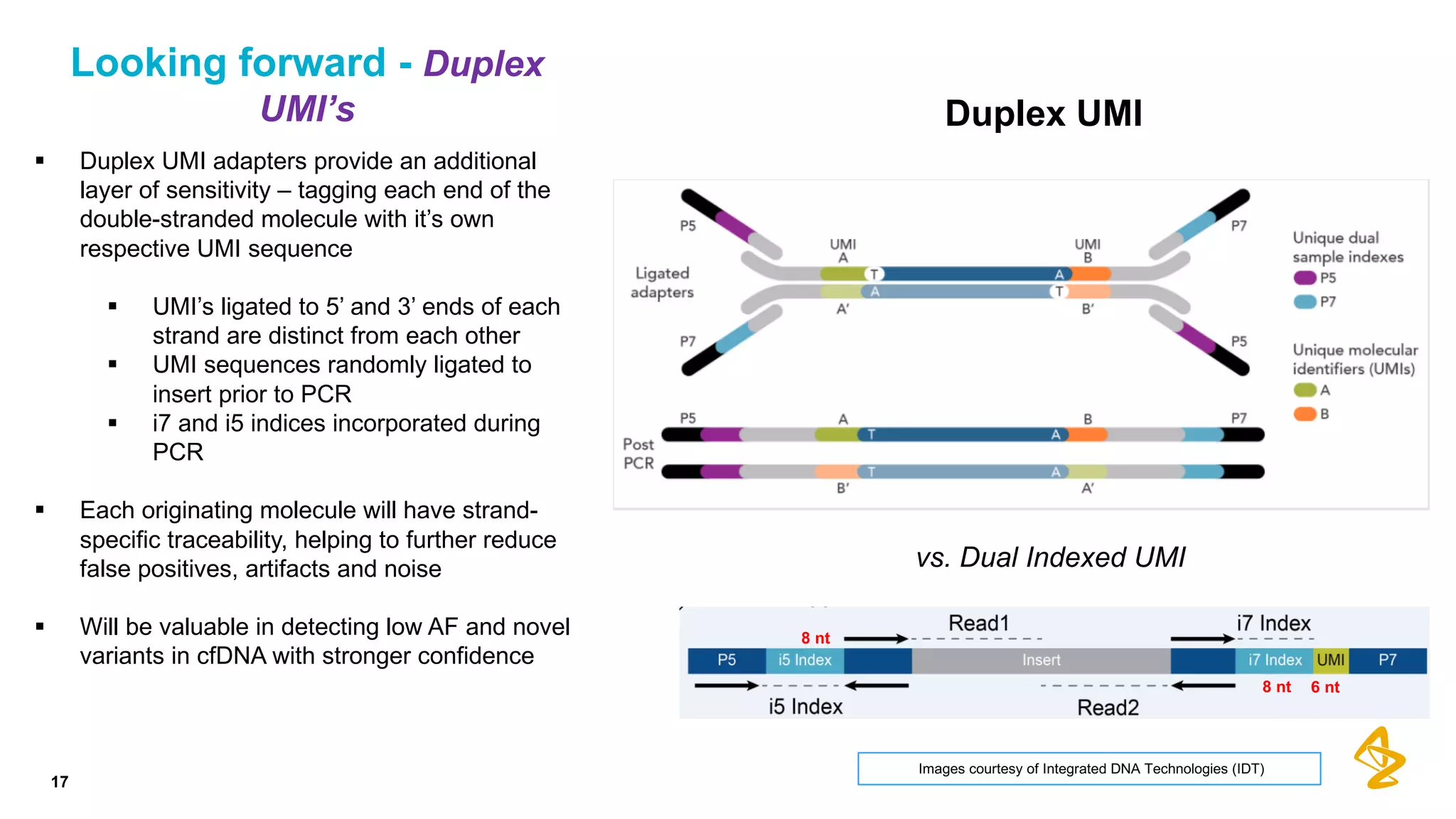




The document discusses advances in next-generation sequencing (NGS) and the use of unique molecular identifiers (UMIs) to enhance the detection of circulating tumor DNA (ctDNA) in plasma samples, significantly improving the sensitivity and specificity of mutation detection in oncology. It highlights the transition from traditional tissue biopsies to liquid biopsies, emphasizing the benefits and challenges of analyzing ctDNA for personalized medicine. Furthermore, it examines the application of machine learning for confident variant calling and the potential future impact of duplex sequencing technology on clinical trial projects.























































































