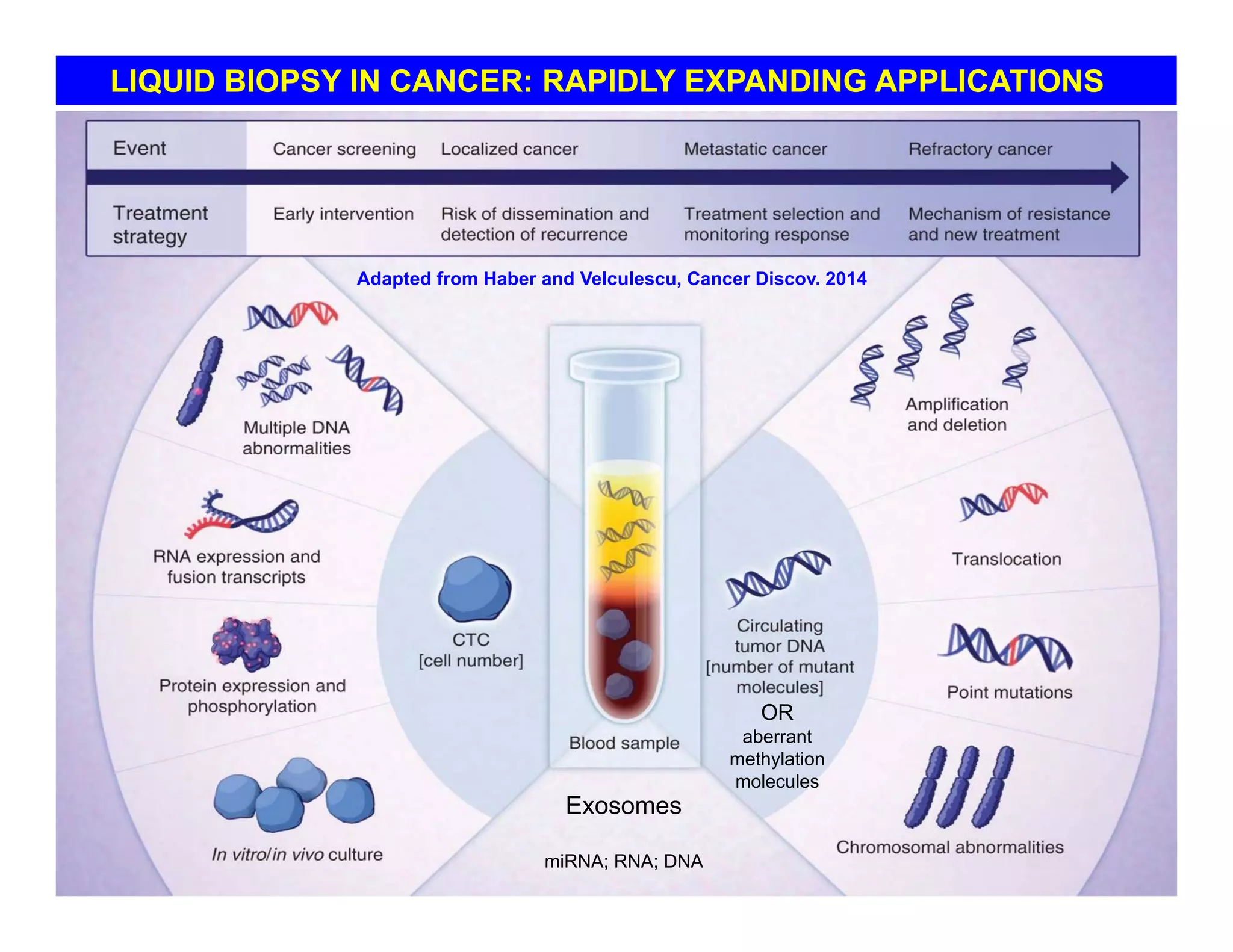
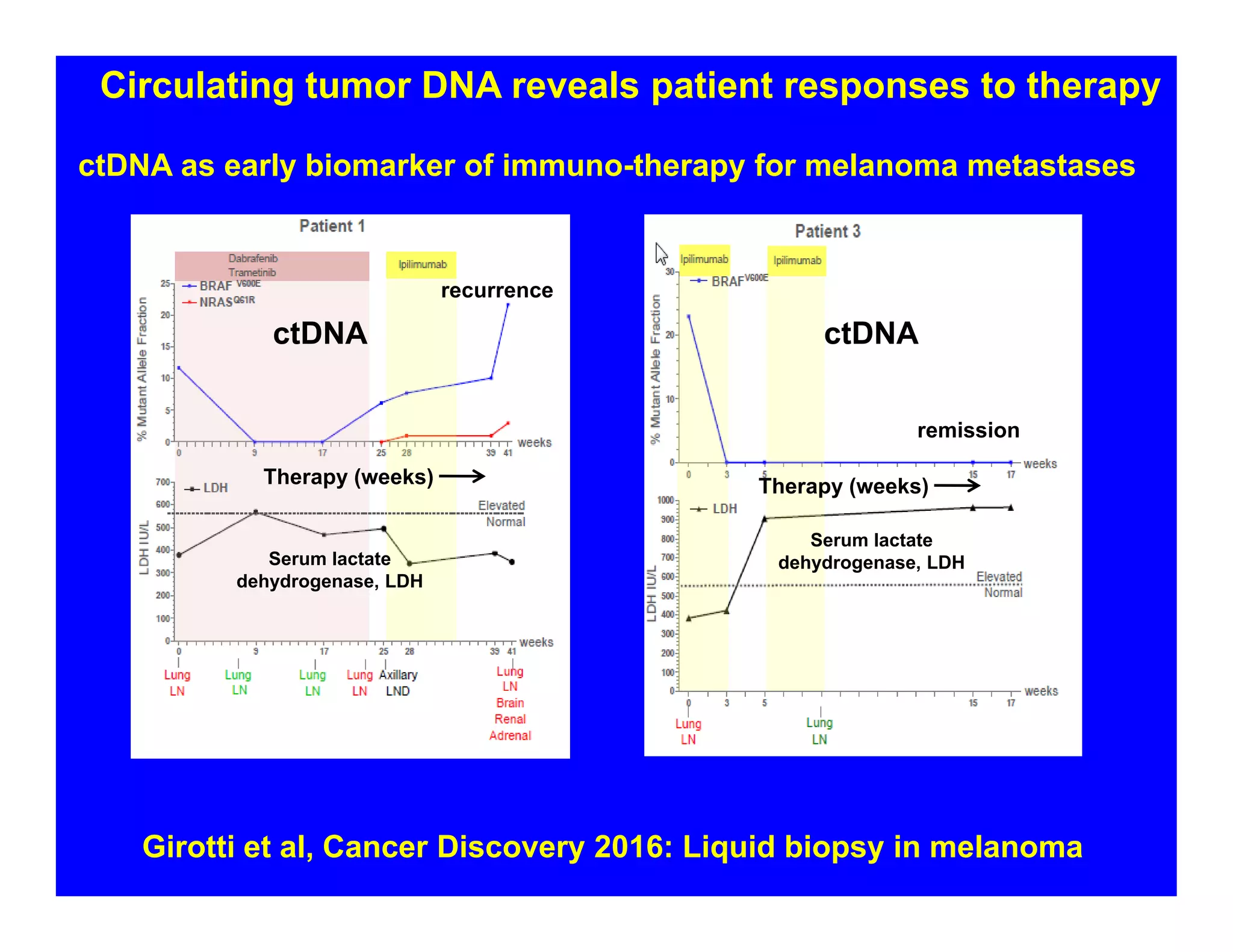
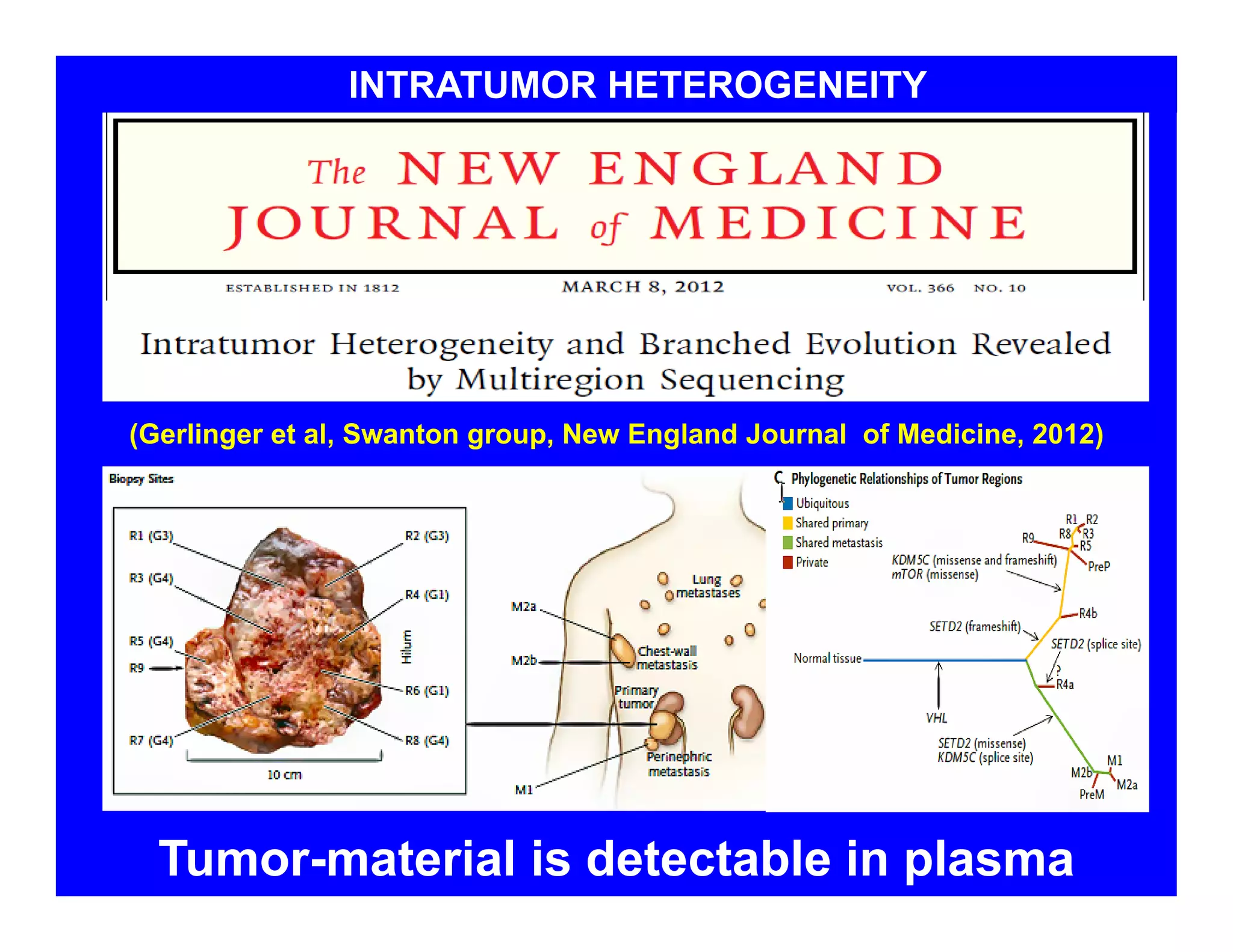
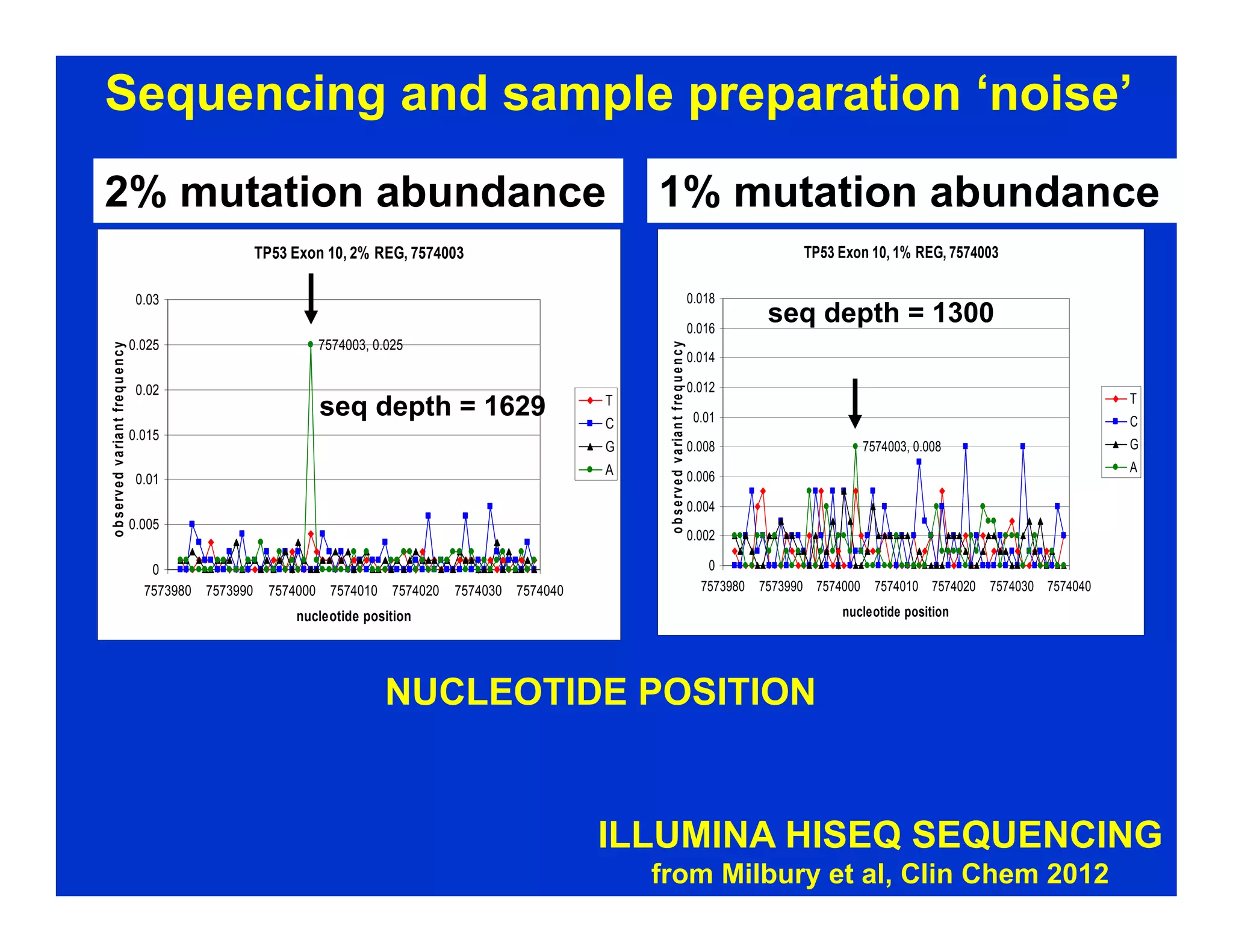
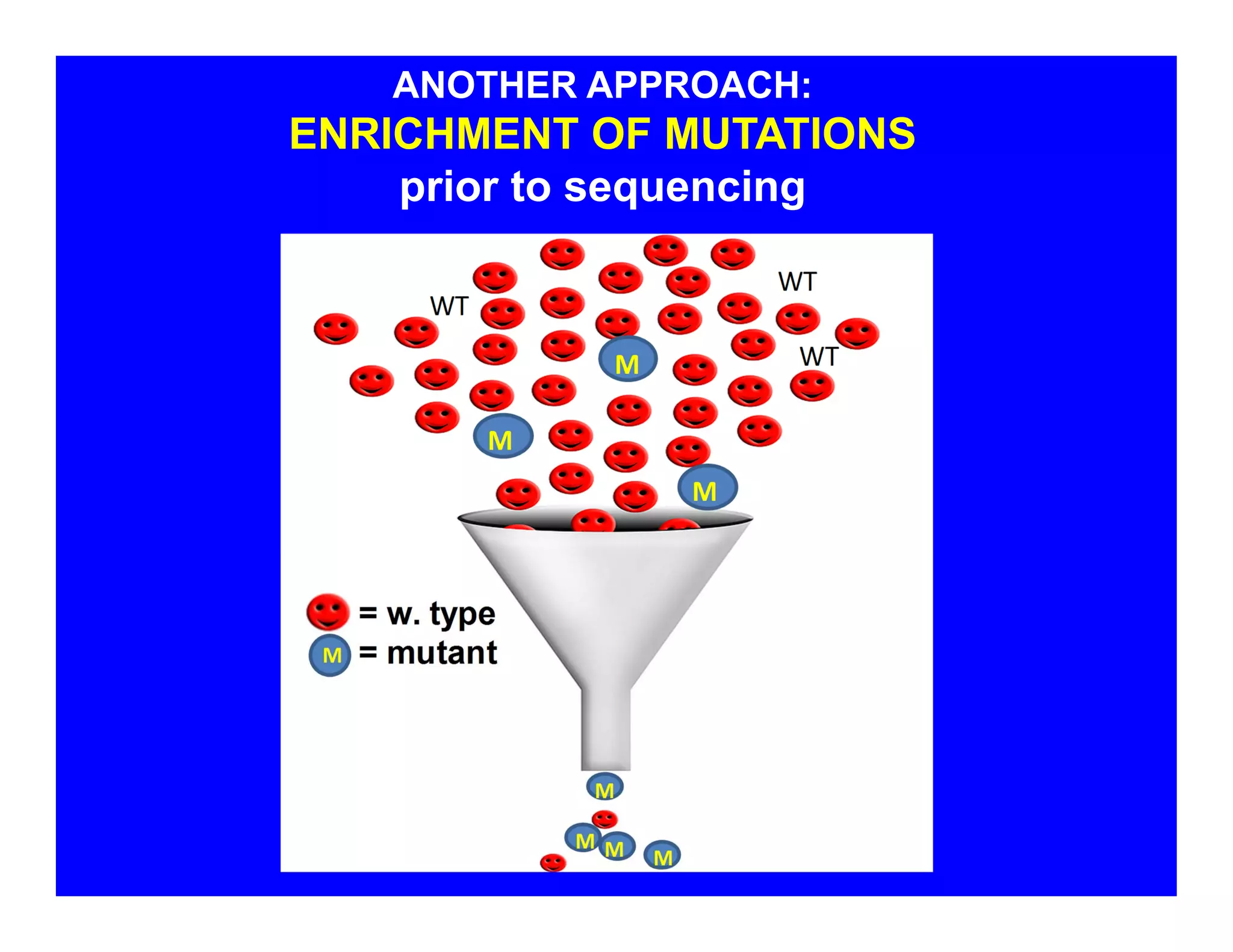
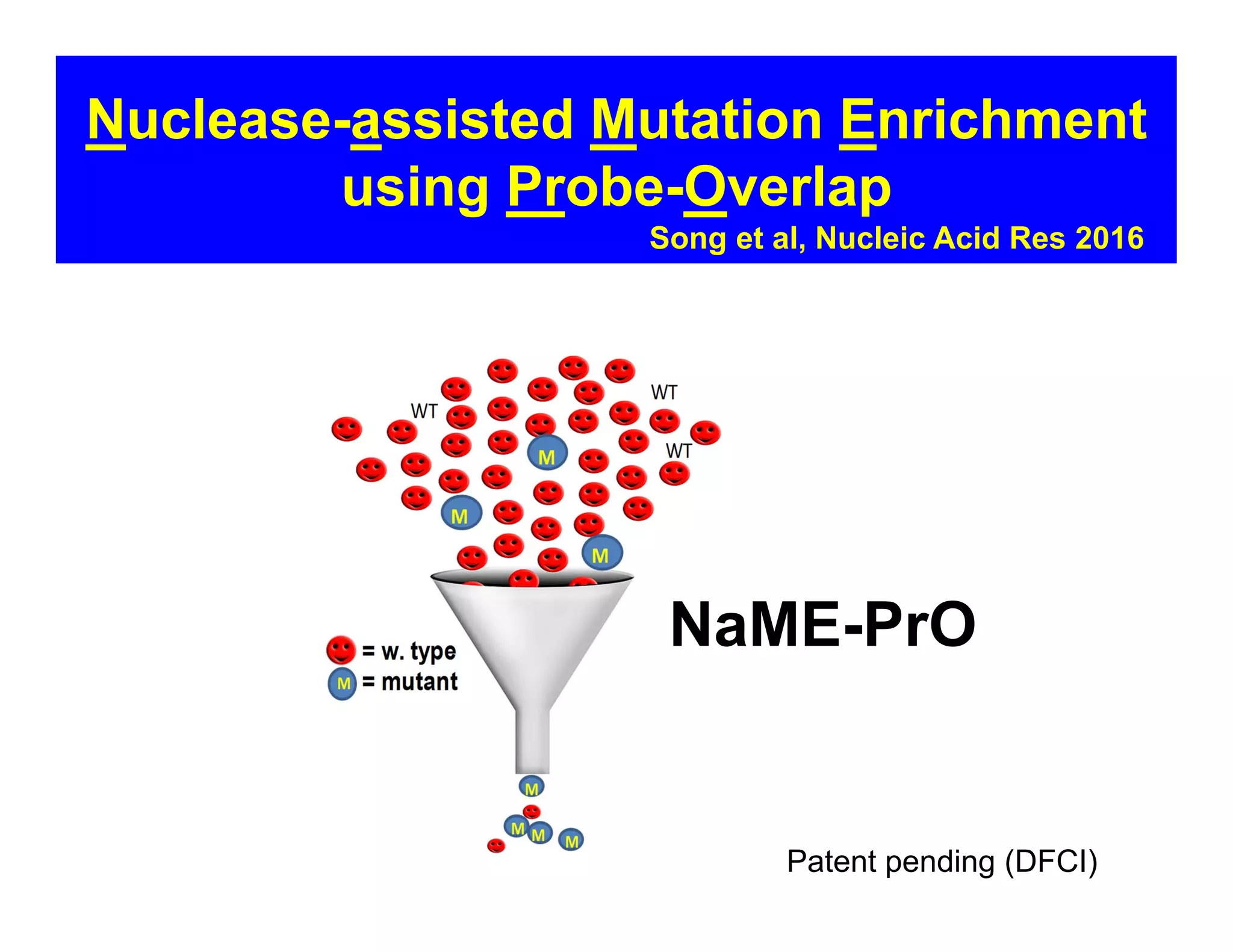
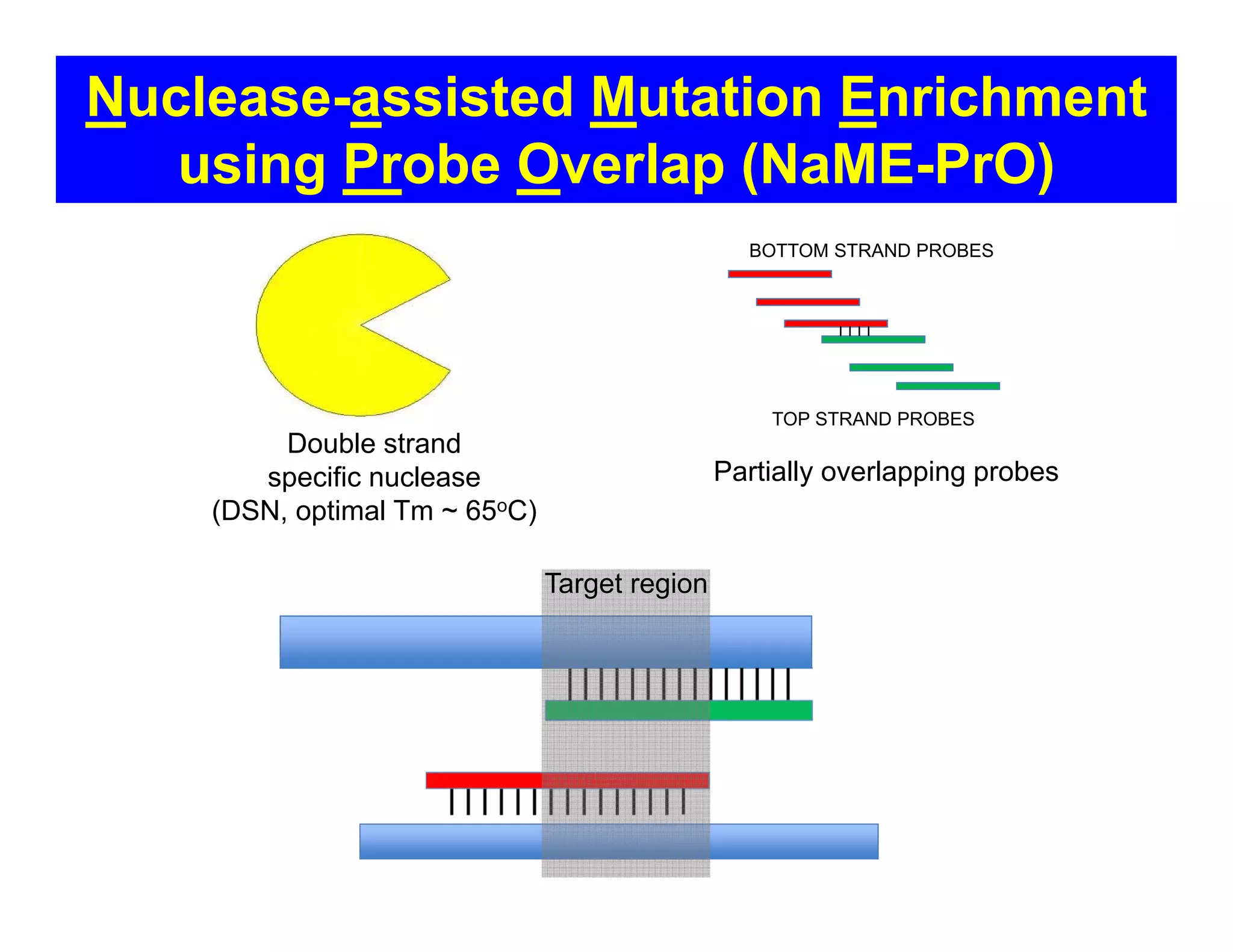
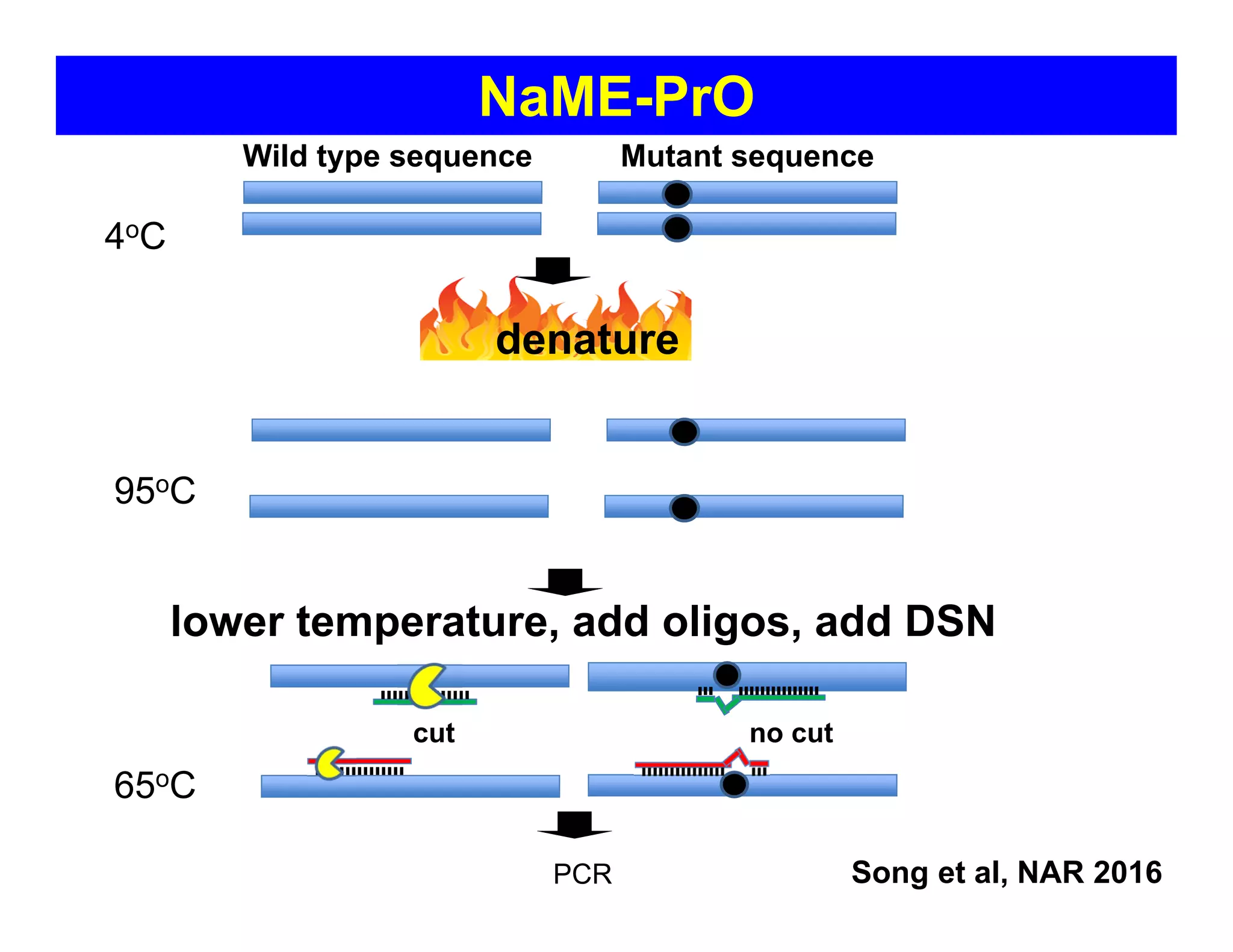
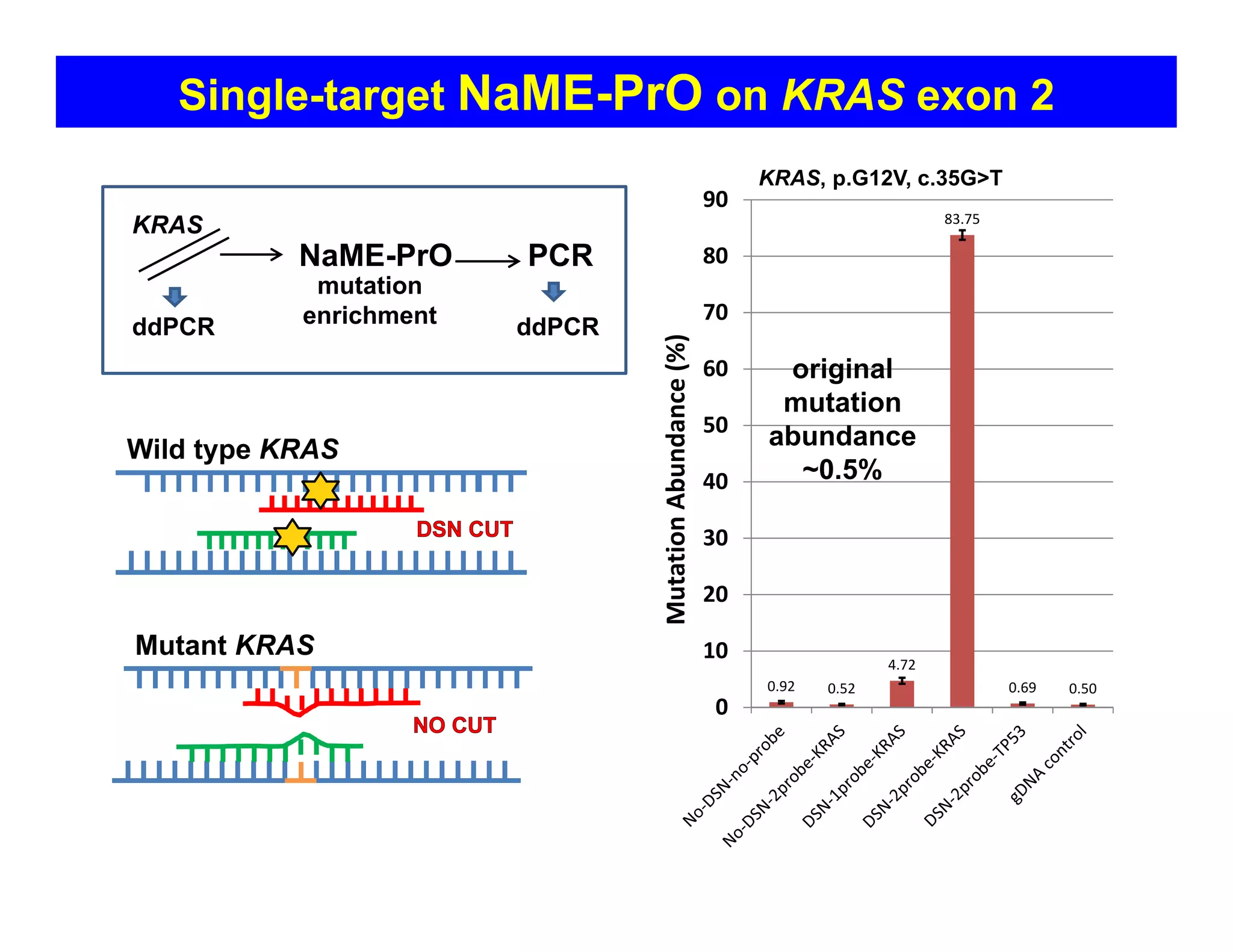
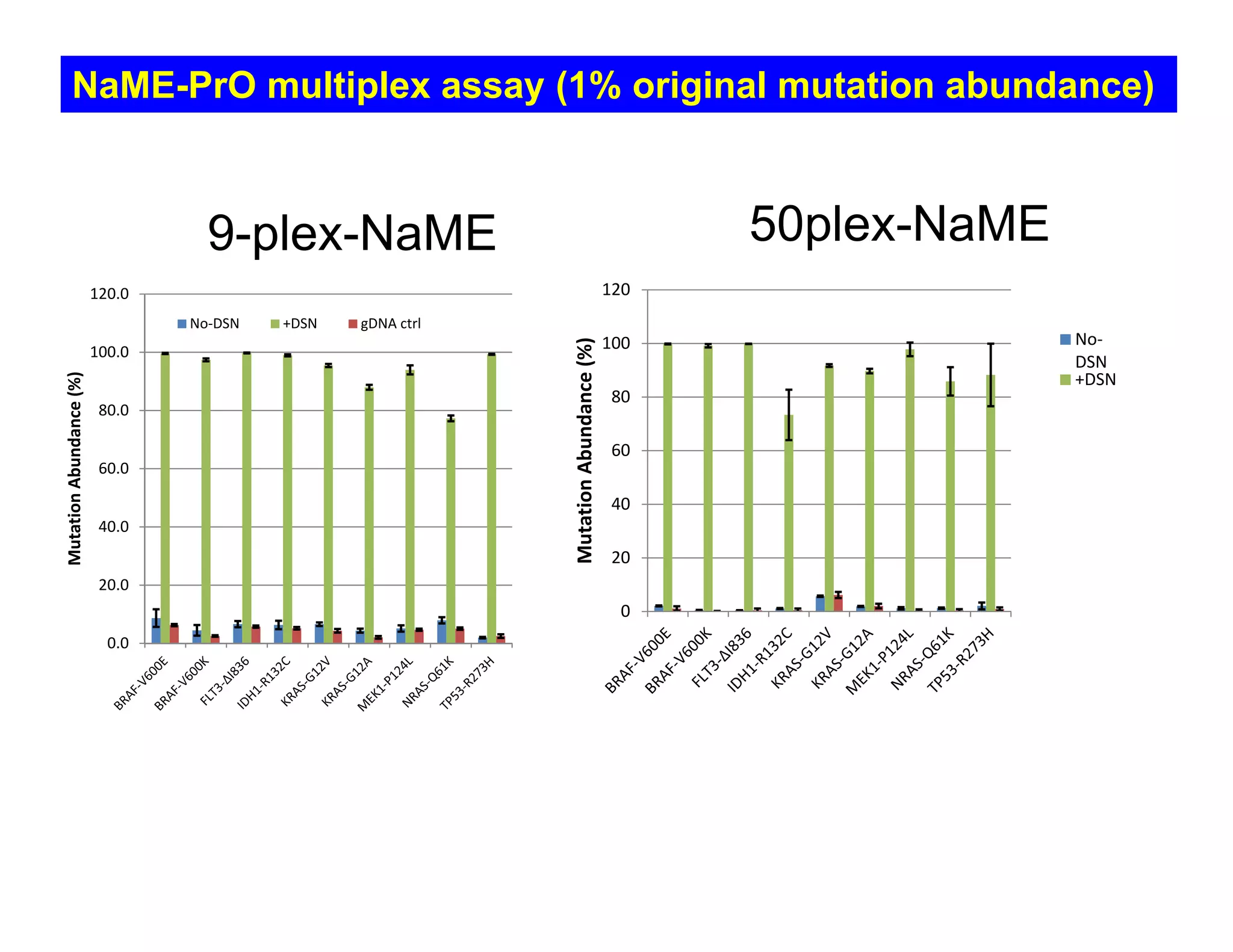
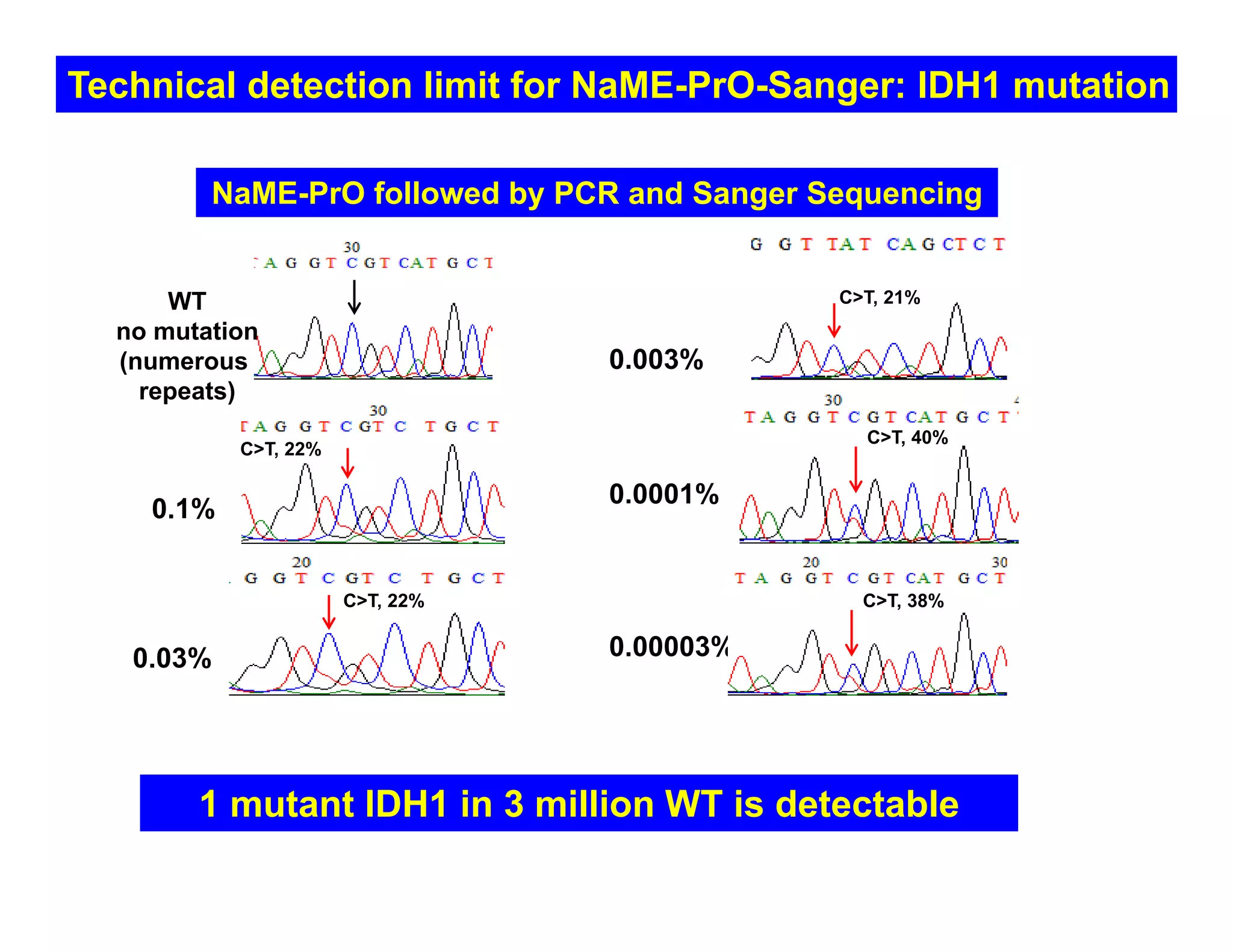
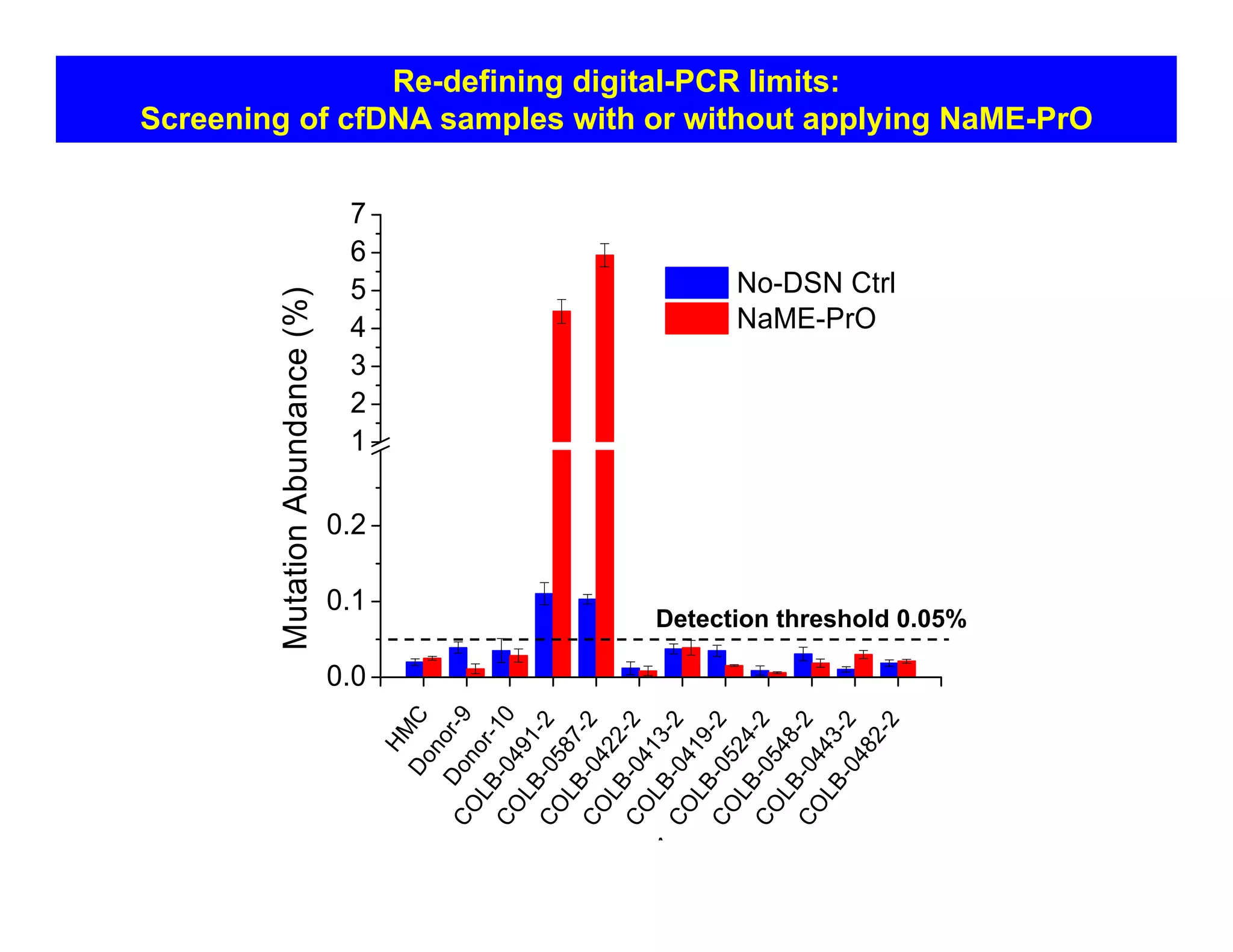
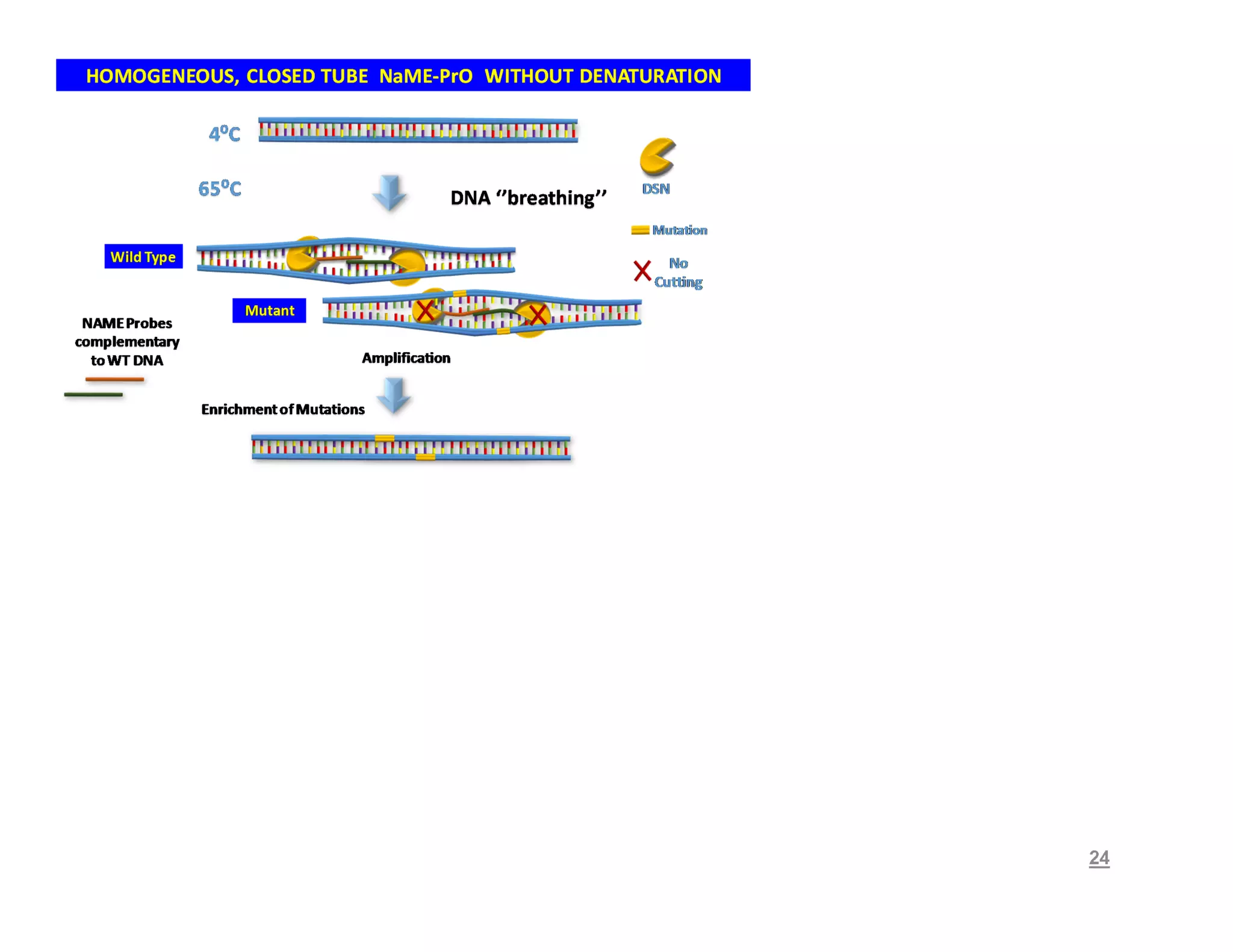
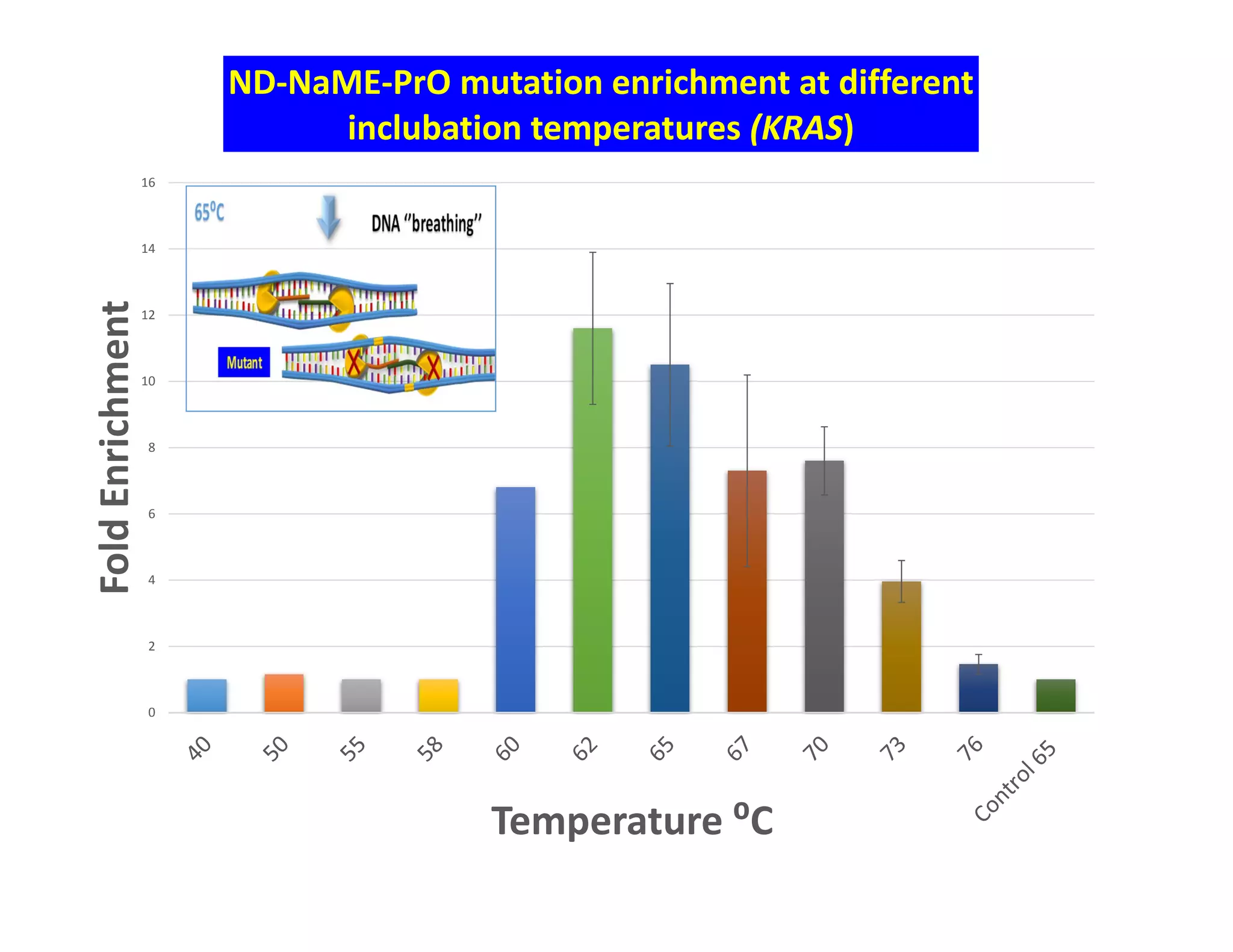
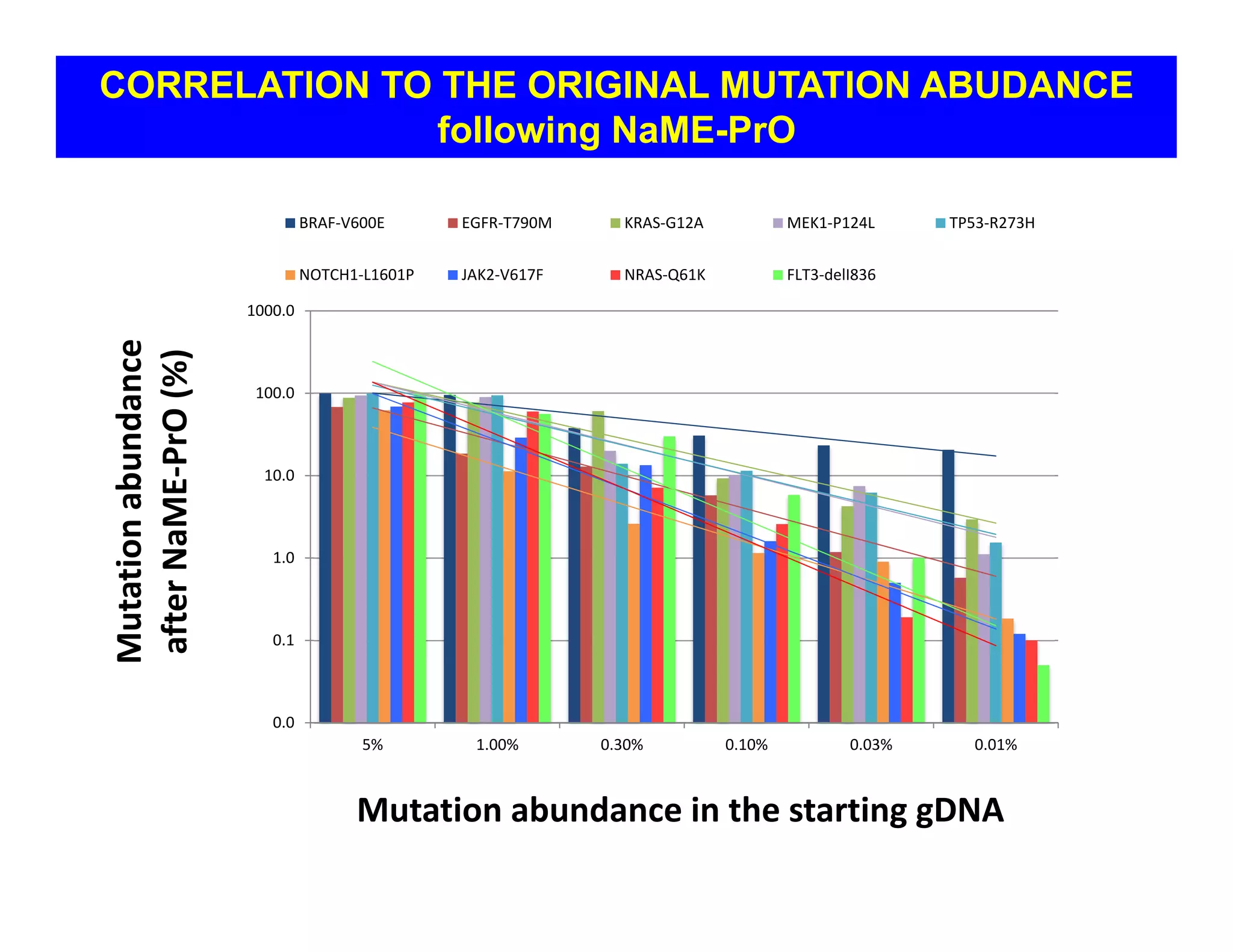
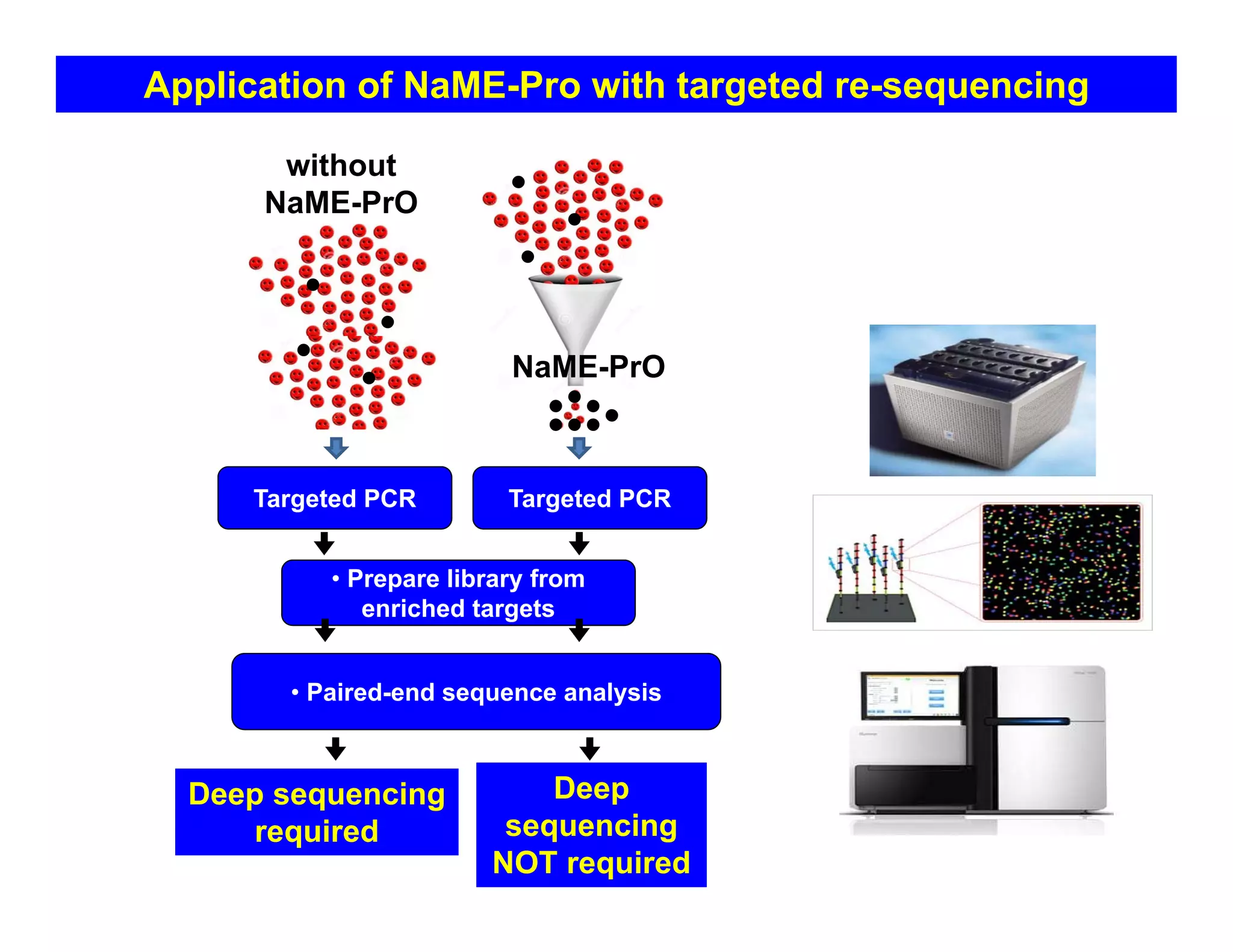
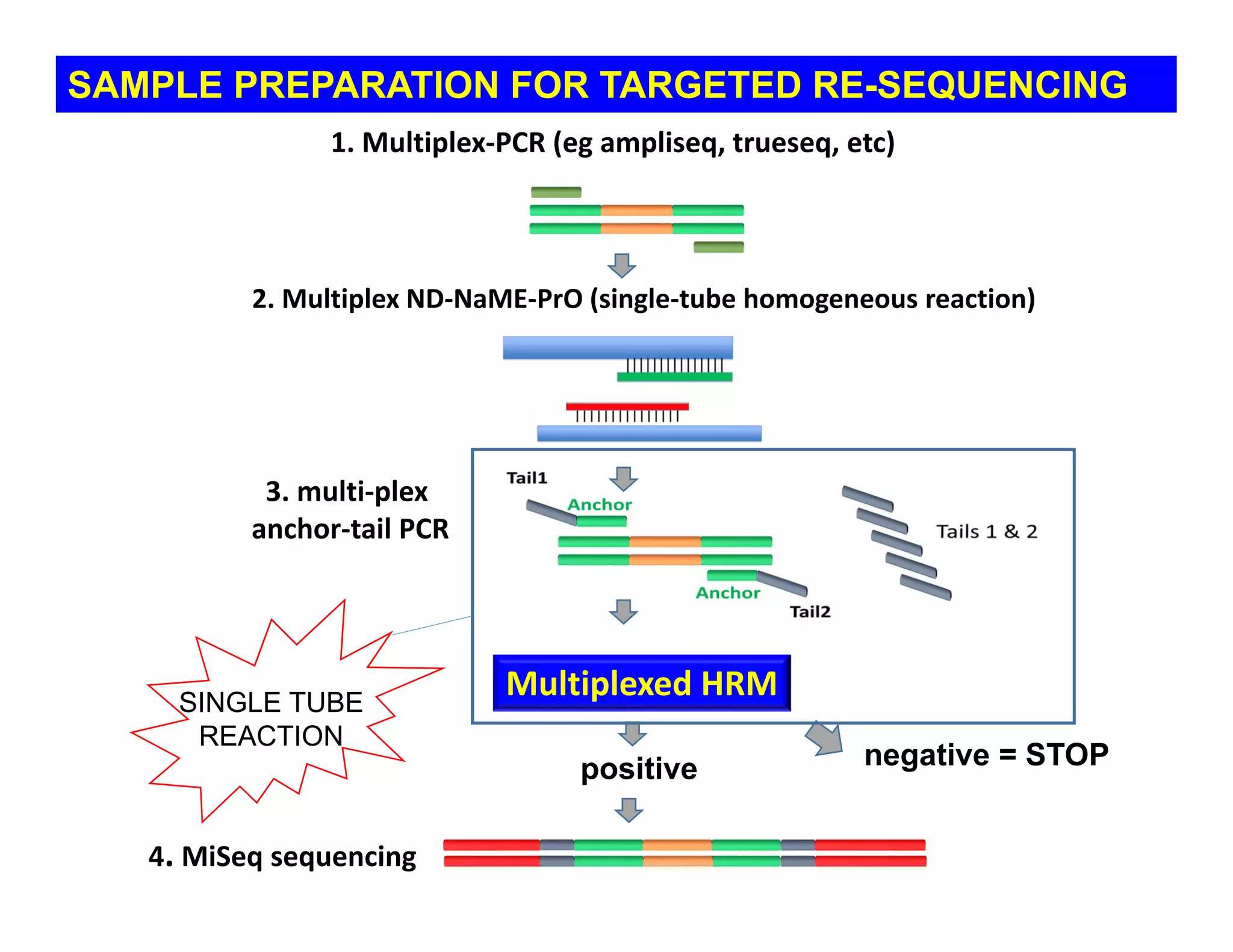
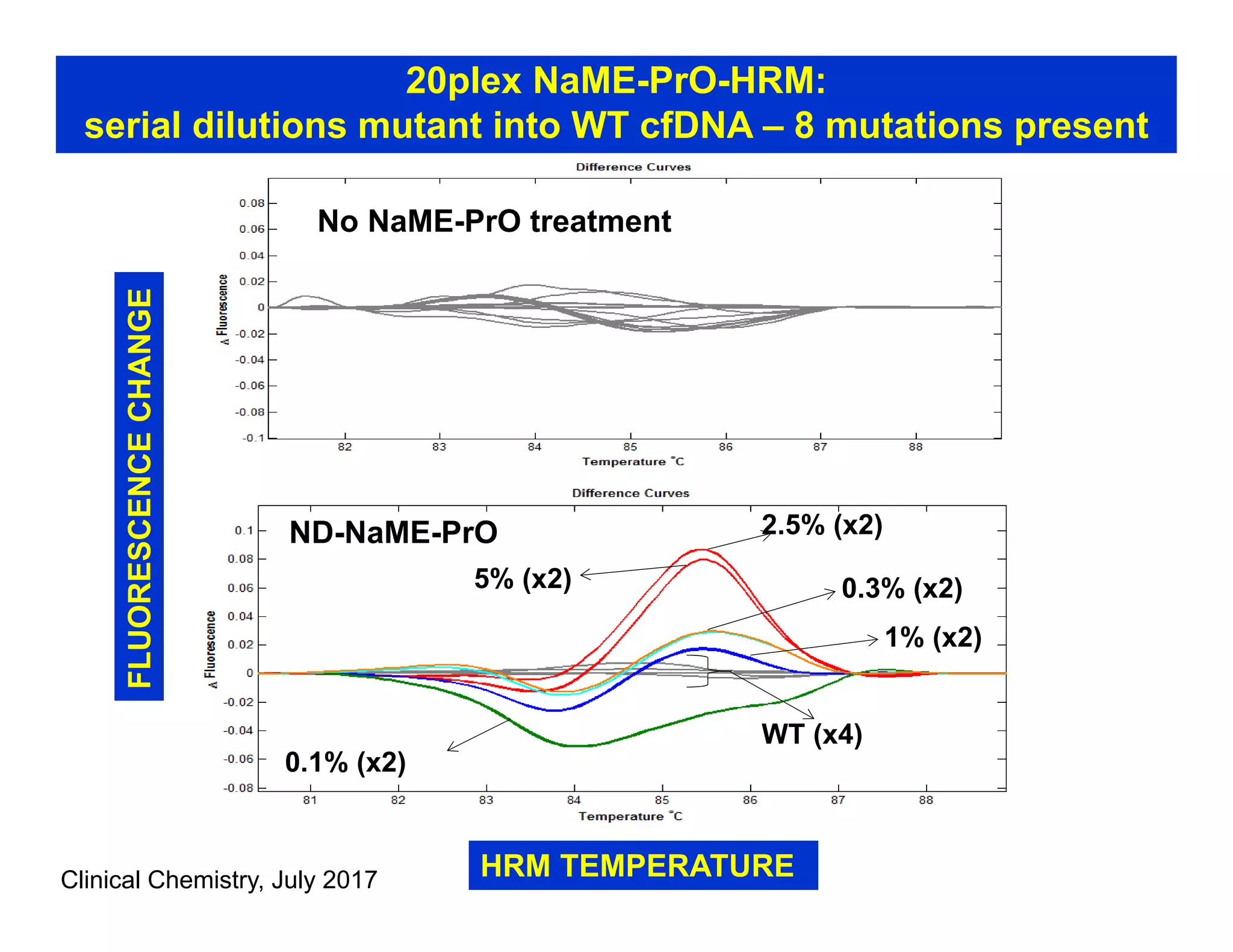
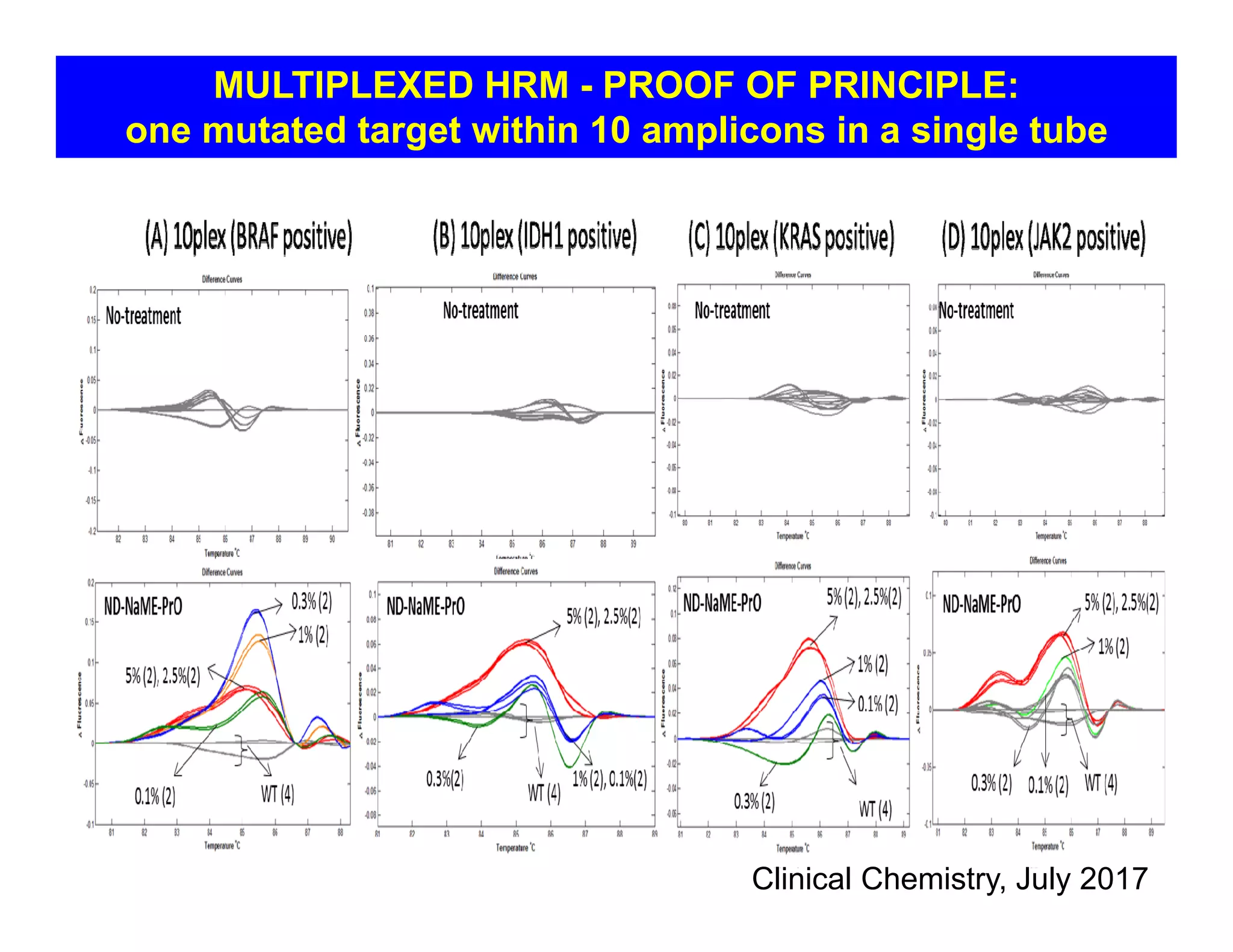
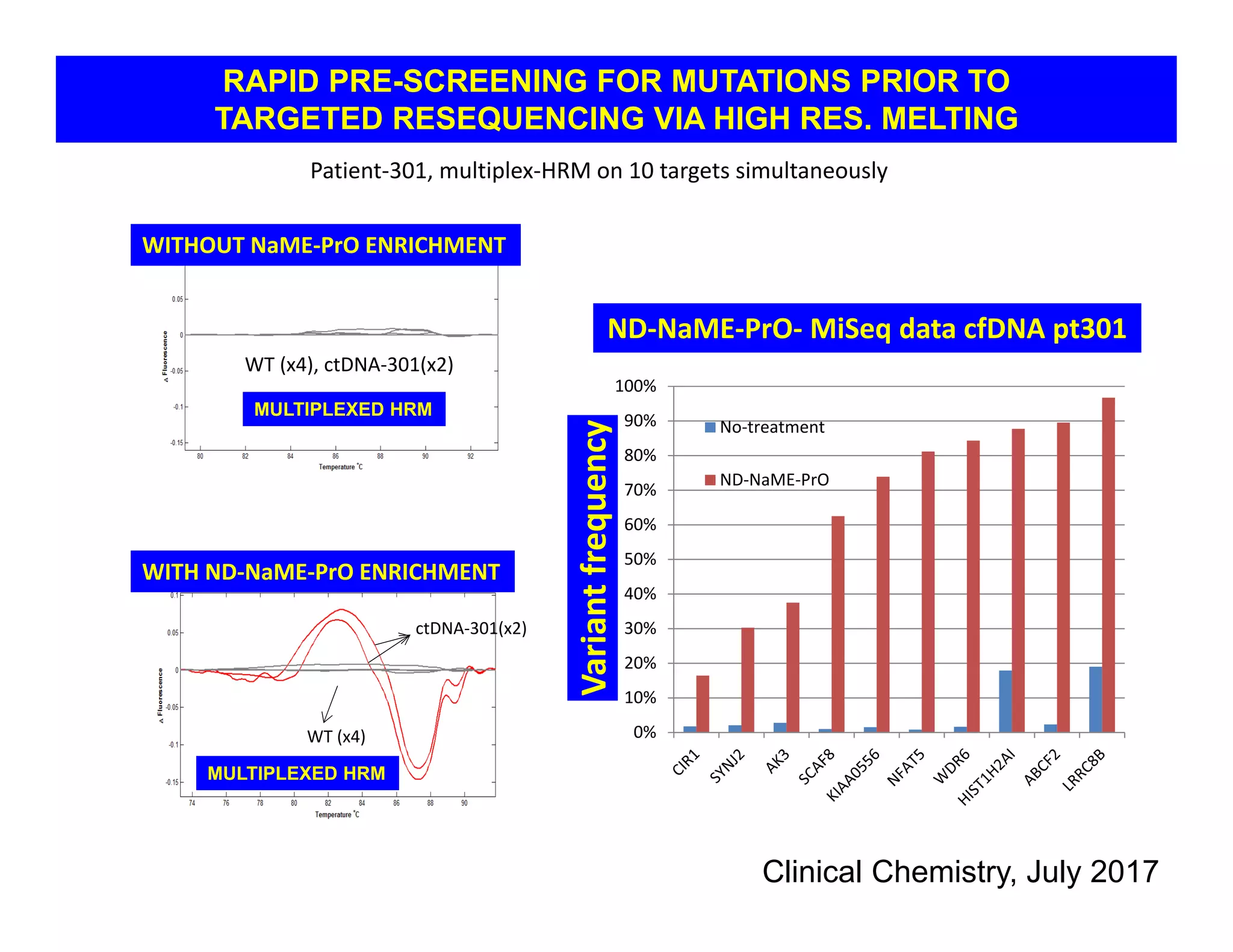
The document discusses advancements in liquid biopsy techniques, particularly focusing on mutation detection through multi-step real-time PCR and high-resolution melting (HRM) analysis. It emphasizes the necessity of targeted re-sequencing for effective cancer biomarker screening and highlights the challenges of sequencing noise and depth in low-abundance mutations. The paper also presents methodologies like the Name-Pro technique, which enhances mutation enrichment, potentially streamlining next-generation sequencing sample preparation for clinical oncology applications.