T-coffee provides concise summaries in 3 sentences or less that provide the high level and essential information from the document.
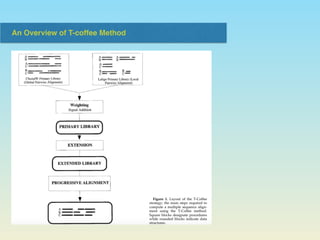
The document describes the T-coffee multiple sequence alignment method. It combines information from global and local alignments, takes reference from other sequences, and extends the primary library through a triplet approach. Validation using BaliBase shows the extended library performs better than primary libraries, and T-coffee outperforms other methods on core region and complete alignment tests. However, T-coffee does not always outperform other methods for all specific cases.








![Primary Library of Alignments (Global and Local)
Library— a set of pairwise alignments between all of the sequences to be aligned, and in a
sequence-to-sequence-position-pair speci8ic weight list form. a library can be stored as a N*N
lower (or upper) triangular matrix where main diagonal can be ignored, and each entry is a
weight list. In other word, a list of weighted pairwise constraints. The primary library of global
alignment for a sequence set is denoted by AG and the primary library of local alignment for a
sequence set is denoted by AL. A* is referred to either AG or AL.
Now suppose we have a sequence set with size N(N refer to the number of sequences in the set) ,
the total number of sequence pairs for the sequence set is N*(N-1)/2.
We can use Ai to denote the ith sequence(item) in the sequence set A. So that in matrix A* we can
know entry A*ij where contain the information from the pair of alignment the entry denotes. Before
to generate global alignment AG or local alignment AL, we should 8irst do all possible pairwise
alignments using global alignment method or FASTA local segments match method(Lalign)
*When we do local pairwise alignment, by default, we choose ten top-scoring non-intersections local
alignment from each pair of alignment. So the number of segments derived from an alignment is very
likely less than 10 (simply because there are no so many qualified matched segments) and could be 0.
After the pairwise alignment, we derived a list of pairwise residue matches for each entry of A*. And Xm
denote the mth position in a certain sequence Ai. So the list in an entry can be denoted by (Xn Xm)|A*ij.
Finally, we assign a weight to each pairwise residues match in all lists directed by all entries in A*,
and the weight equal to percentage identity of the alignment of Ai to Aj where the pairwise residue
match is derived from. W[(Xn, Xm)|A*ij] = P.I.(A*ij). The weight is also referred as constraint.](https://image.slidesharecdn.com/t-coffeealgorithmdissection-160826052714/85/T-coffee-algorithm-dissection-11-320.jpg)

![Combination of the Libraries: Addition
Pooling the ClustalW and Lalign primary libraries in a
simple process of addition:
AGL$=$AG+AL$
W[(Xn, Xm)|AGLij] = W[(Xn, Xm)|AGij]+ W[(Xn, Xm)|ALij]
If W[(Xn, Xm)|A*ij] is not recorded in A*, assign 0 to it.
Then entry AGLij can be regarded as a ‘sparse’ list with L(i)*L(j) number of
key-value pairs (a lot of values are 0). L(i) denote the length(or number of
residues) of sequence Ai.](https://image.slidesharecdn.com/t-coffeealgorithmdissection-160826052714/85/T-coffee-algorithm-dissection-13-320.jpg)


![Extending the library: Triplet approach
W(A(G), C(?)) W(A(G), C(?))consider seqC
consider seqD W(A(G), D(?)) W(A(G), D(?))
For W(A(G), B(G)) E[W(A(G), B(G))]=W(A(G), B(G))+%d=88+77
If C(?) == C(?): get %(min) of W(A(G), C(?))=77
W(A(G), D(?))=100else %(0)
v
v
Sometimes we will get better alignment
If we don’t strictly follow the guide tree.
That is why we take inference from other
sequences when align two sequences
following the guide tree.
Iterative method achieve this goal by
modifying guide tree in a heuristic manner.
e.g. MUSLE](https://image.slidesharecdn.com/t-coffeealgorithmdissection-160826052714/85/T-coffee-algorithm-dissection-16-320.jpg)
![Extending the library: Let’s code this process
Note the library extension operator as AE and notice that it is not a library that can
be added to A* because it is a function of A*. AE(A*)= A*E.
def AE (A*):
for i=1, i++, i<=N
for j=i+1, j++, j<=N // go through A*ij: C(2,N)
for m=1, m++, m<=L(Ai)
for n=1, n++, n<=L(Aj) //go through all
constraints in the matrix entries: L^2
E=0, for k of each Ak belonging to A-Ai-Aj
a = get_position(m i k a)
b = get_position(n j k b)
if a == b // to find consistent
residues in other sequences supporting match of
Xm|Ai and Xn|Aj: 2L
e1 = W[(Xm, Xa)|A*ik]
e2 = W[(Xn, Xb)|A*jk]
E +=min{e1, e2} // get extension
weight
W[(Xm,Xn)|A*ij]+= E // A*E
def get_position(m i k a):
for n=1 n++ n<=L(Ak)
if W[(Xm,Xn)|A*ik] != 0
add n to a // find the possible consistent
position in Ak: L(Ak)
return a
C(2,N)* (L^2)*L=O(N^2*L^3)](https://image.slidesharecdn.com/t-coffeealgorithmdissection-160826052714/85/T-coffee-algorithm-dissection-17-320.jpg)


![Progressive Alignment Strategy
Given the Column n
C
C
C
T
+
T
T
T
! !, !!
!!!
!
!!!!!
!!
!
= !"#$!%#_1(!1)!!
! !, !!
!!!
!
!!!!!
!!
!
= !"#$!%#_2(!2)!!
C
C
C
T
T
T
T
+
C
C
Don’t need to align pairs of residues within existing column of
alignment , only consider weights of matched pairs of residues
between existing column:
!!
!!! [!"#!(!), !"#!(!)]!
!!!!!
!!
!
∗ !!
!
= !"#$!%#_3(!3)!!
average_1’=a1+a2+a3
average_2’
Within Within Between
average_3’](https://image.slidesharecdn.com/t-coffeealgorithmdissection-160826052714/85/T-coffee-algorithm-dissection-20-320.jpg)