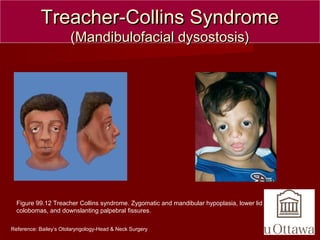
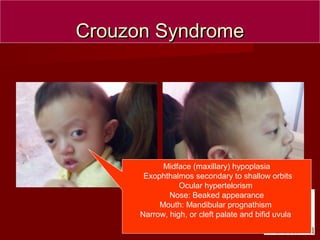
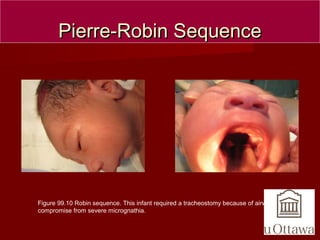
The document discusses several craniofacial anomalies including DiGeorge Syndrome, Treacher-Collins Syndrome, Apert Syndrome, Crouzon Syndrome, Branchiootorenal Syndrome, Down Syndrome, Goldenhar Syndrome, and Pierre-Robin Sequence. It provides the genetic causes, characteristic features, and figures to illustrate each condition. Craniosynostosis and cloverleaf skull syndrome are discussed in more detail, with craniosynostosis defined as the premature fusion of cranial sutures, which can be primary, secondary, or syndromic, and the roles of specific sutures explained.