Downloaded 1,086 times






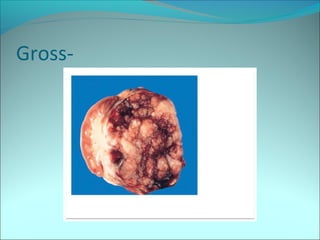

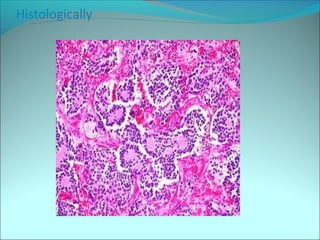
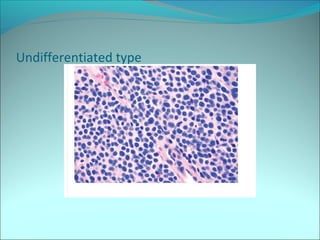
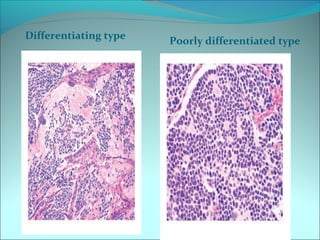

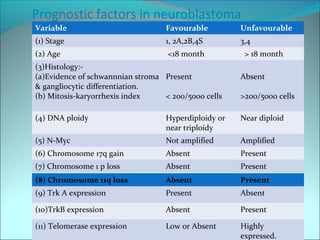



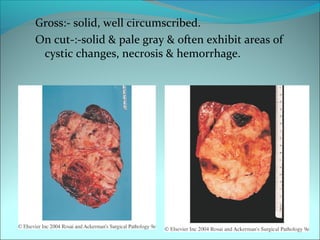


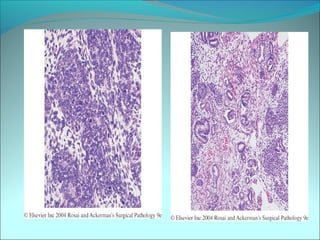
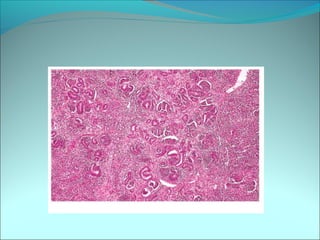
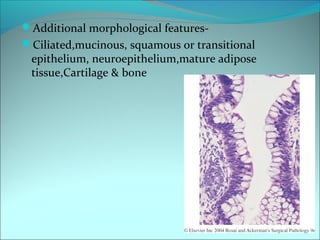
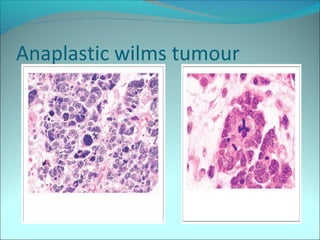



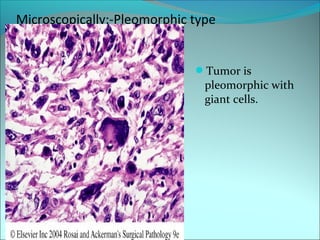

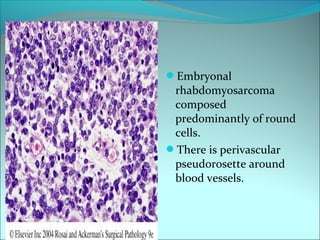
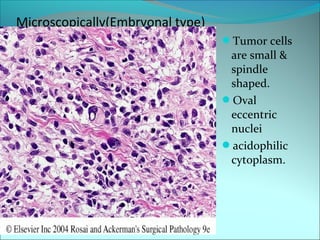
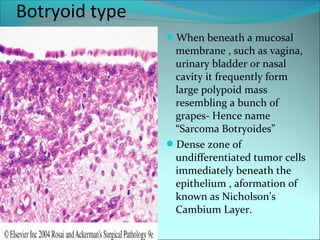

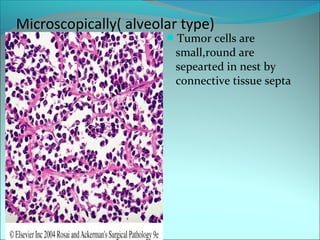
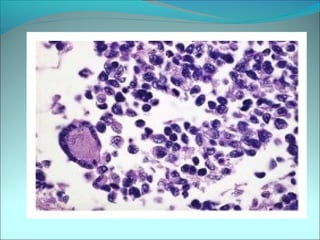






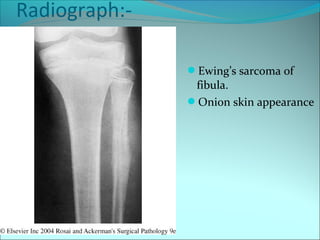
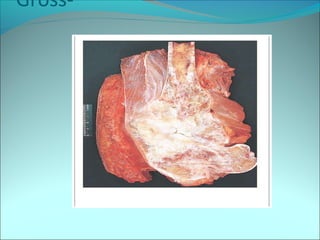
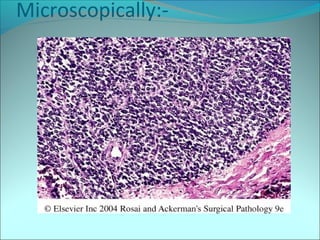
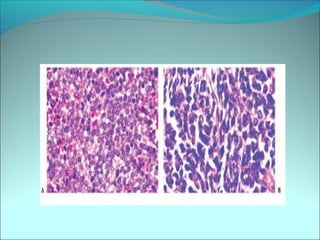
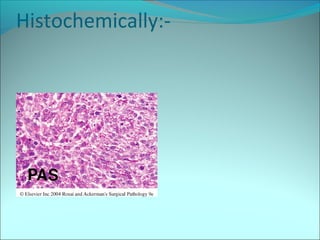



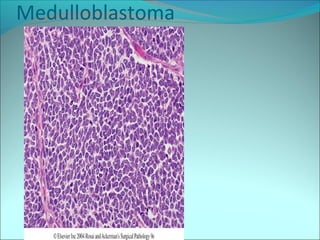
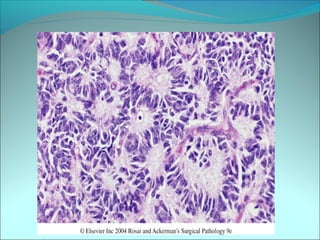
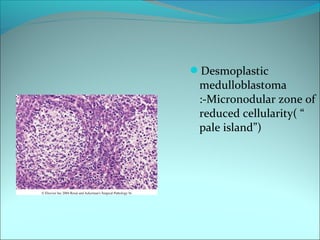
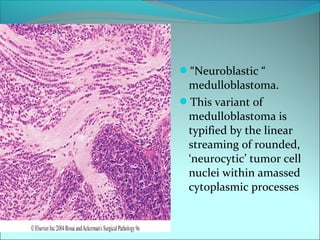
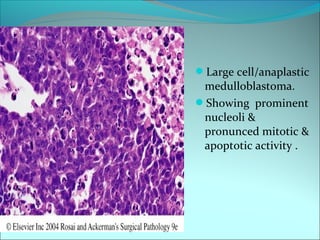

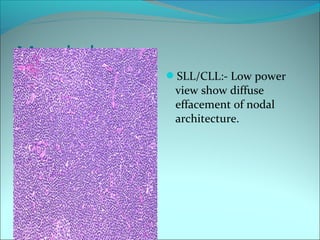
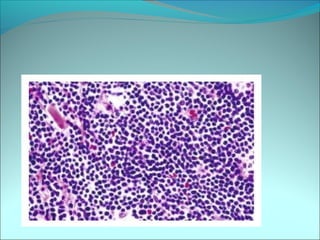

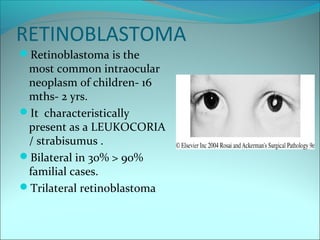


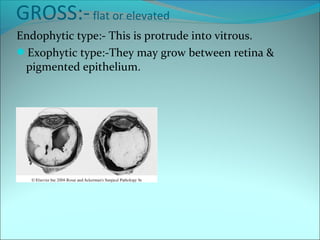
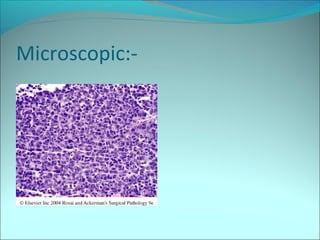
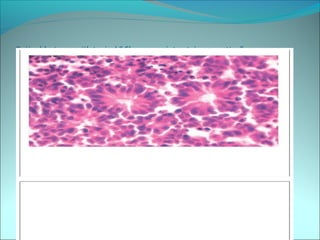
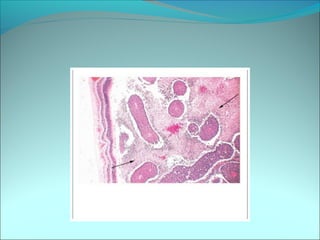




The document discusses several pediatric neoplasms that appear as small round blue cell tumors due to their primitive histological features. These include neuroblastoma, Wilms tumor, rhabdomyosarcoma, Ewing's sarcoma, medulloblastoma, retinoblastoma, and lymphoma. For each tumor, the document outlines characteristics such as common age of diagnosis, clinical features, histopathological appearance under the microscope, immunohistochemistry profiles, genetics where relevant, and important prognostic factors. Differential diagnosis of these small round blue cell tumors in children is provided for accurate diagnosis and treatment.



























































































