Download to read offline


![What is NJ Method?
● A method called the neighbor-joining method was proposed for
reconstructing phylogenetic trees from evolutionary distance
data.
● The NJ method was developed by Saitou and Nei (1987).
● The principle of this method is to find pairs of operational
taxonomic units (OTUs [ =neighbors]) that minimize the total
branch length at each stage of clustering of OTUs starting with
a starlike tree.
● The input is the ‘n’ number of taxa.
● The output is an unrooted tree with branched](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-3-2048.jpg)




![Step 2: Now we calculate a new distance matrix using for
each pair of OTUs the formula:
M(ij)=d(ij) - [r(i) + r(j)]/(N-2) or in the case of the pair A,B:
A B C D E
B -13
C -11.5 -11.5
D -10 -10 -10.5
E -10 -10 -10.5 -13
F -10.5 -10.5 -11 -11.5 -11.5
M(AB)=d(AB) -[(r(A) + r(B)]/(N-2) = -13](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-8-2048.jpg)
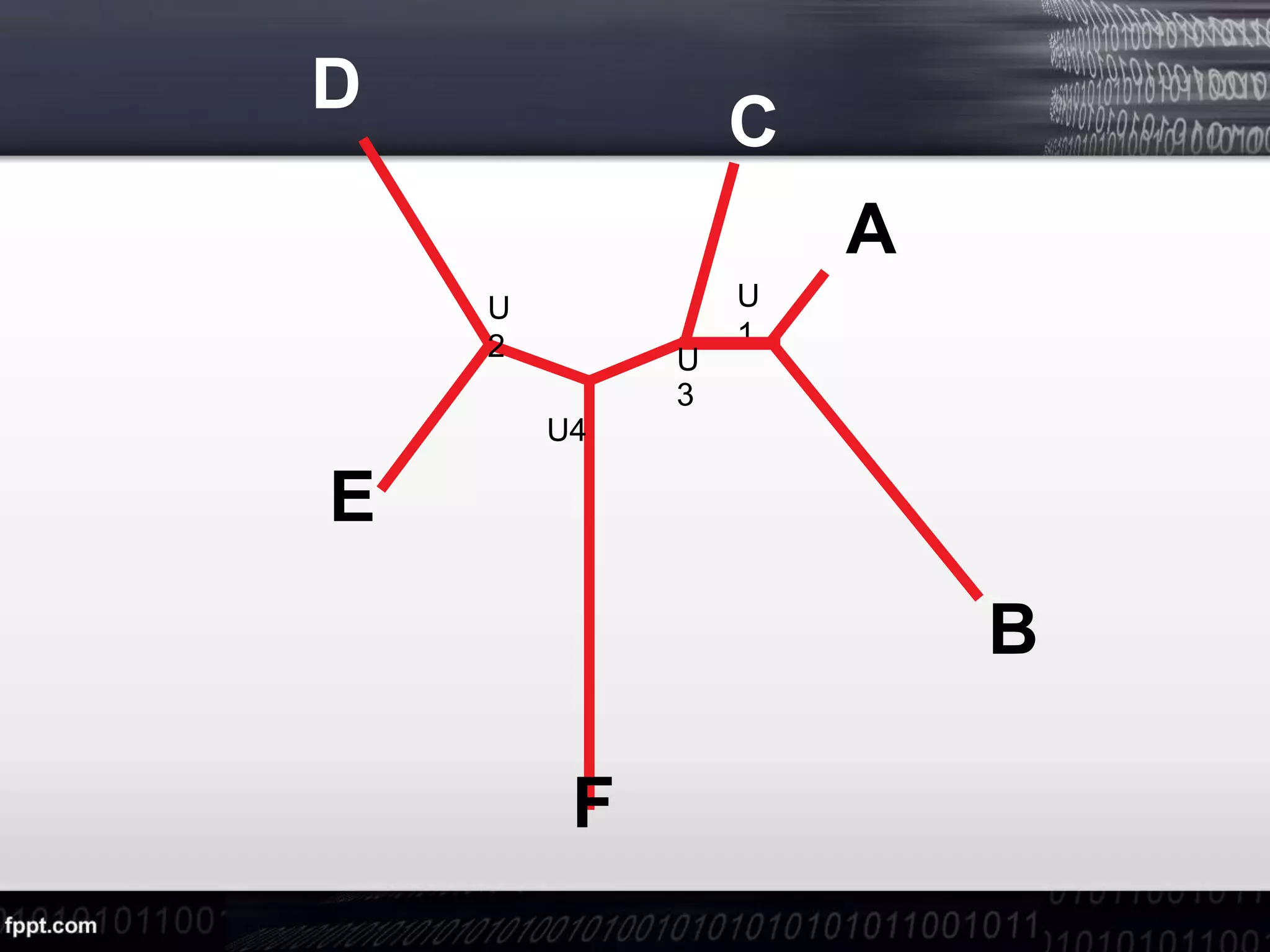
![Step 3:Now we choose as neighbors those two OTUs for which Mij is the
smallest. These are A and B and D and E.
Let's take A and B as neighbors and we form a new node
called U. Now we calculate the branch length from
the internal node U to the external OTUs A and B.
S(AU1) =d(AB) / 2 + [r(A)-r(B)] / 2(N-2) = 1 S(BU1)
=d(AB) -S(AU1) = 4](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-9-2048.jpg)


![1. r(U1) = 3+3+7=13
r(C) = 15
r(U2) = 13
r(F) = 21
2. M(ij)=d(ij) - [r(i) + r(j)]/(N-2)
U1 C U2
C -11
U2 -10 -10
F -10 -10 -11
3. S(CU3) = d(CU1) / 2 + [r(C)-r(U1)] / 2(N-2) = 2 S(U1U3)=
d(CU1)-S(CU3) = 1](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-12-2048.jpg)

![1. r(U1) =3+6+5+7=21
r(C) = 24
r(D) = 27
r(E) = 24
r(F) = 32
2. M(ij)=d(ij) - [r(i) + r(j)]/(N-2)
U1 C D E
C -12
D -10 -10
E -10 -10 -12
F -10.66 -10.66 -10.66 -10.66
3. S(DU2) = d(DE) / 2 + [r(D)-r(E)] / 2(N-2) = 3 S(EU2)= d(DU2)-S(DE)
= 2](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-14-2048.jpg)




![1. r(U2) = 8
r(U3) = 6
r(F) = 12
2. M(ij)=d(ij) - [r(i) + r(j)]/(N-2)
3. S(FU4) = d(FU2) / 2 + [r(F)-r(U2)] / 2(N-2) = 5 S(U2U4)=
d(U2F)-S(FU4) = 1
U
2
U
3
U
3
-
12
F -
14
-
12](https://image.slidesharecdn.com/sequenceanalysis-200510134528/75/Sequence-analysis-19-2048.jpg)






The document discusses the neighbor-joining method for reconstructing phylogenetic trees from evolutionary distance data. It was developed by Saitou and Nei in 1987. The method works by finding pairs of operational taxonomic units (OTUs) that minimize the total branch length at each stage of clustering starting with a starlike tree. The input is the number of taxa and the output is an unrooted tree with branches. The document then provides a step-by-step example of applying the neighbor-joining method to calculate and construct a phylogenetic tree from distance data between 6 OTUs.