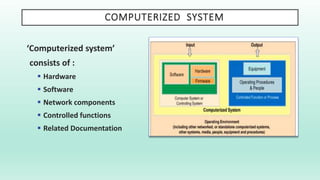
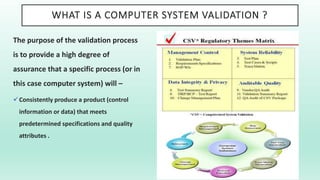
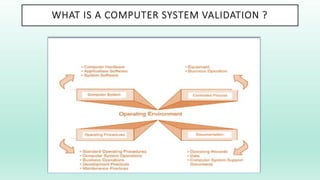
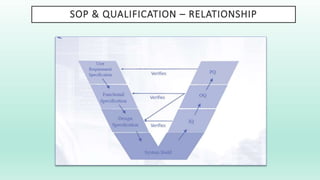
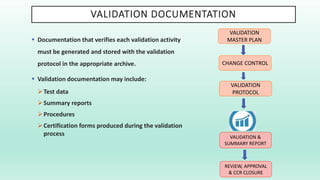
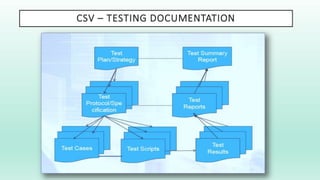
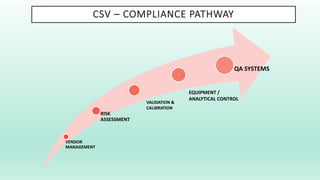
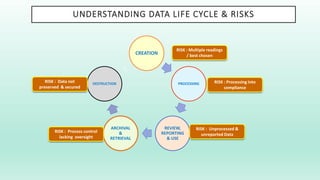
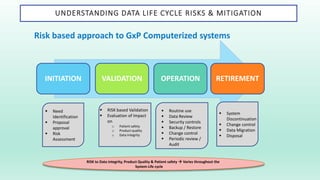
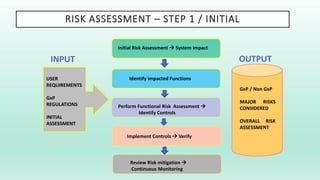
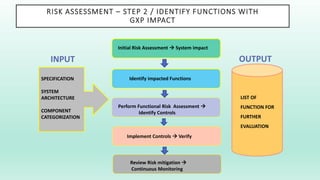
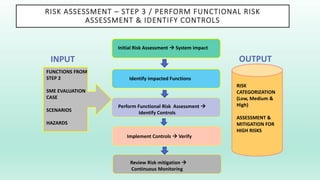
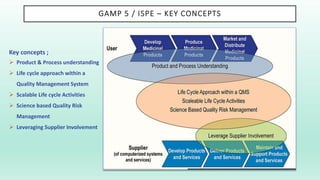
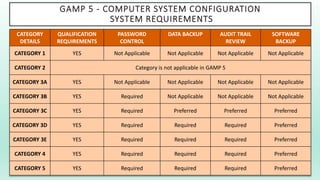
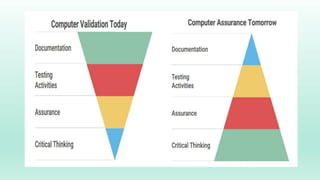
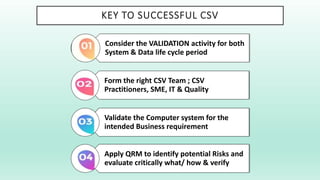
The document outlines a Validation Master Plan (VMP) for the validation of computer systems, specifying requirements for ensuring accuracy, reliability, and performance compliance in regulated environments. It covers the necessary processes, roles and responsibilities, risk assessments, and documentation required for effective validation to ensure patient safety, product quality, and data integrity. Additionally, the document discusses the risk-based approach for computerized system validation and introduces GAMP 5 as a framework for implementing these practices in the pharmaceutical industry.