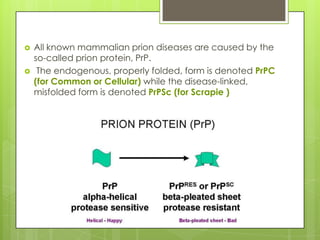
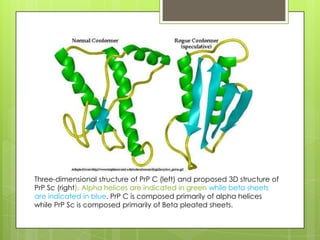
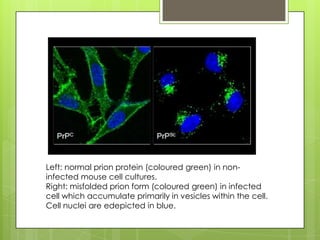
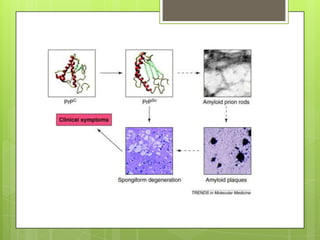
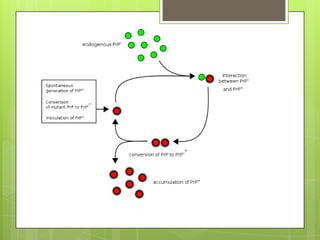
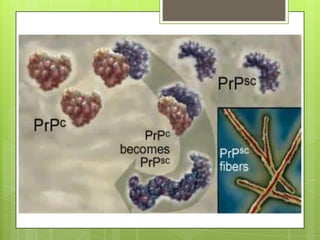
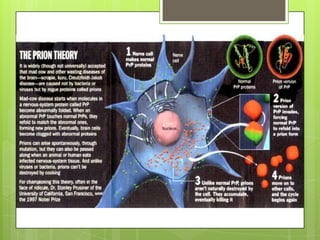
Prion diseases are rare neurodegenerative disorders caused by misfolded prion proteins. They affect both humans and animals. In cattle it is known as bovine spongiform encephalopathy (BSE) or "mad cow disease", and in humans it is known as Creutzfeldt-Jakob disease (CJD). Prion diseases occur when normal prion proteins misfold and induce other prion proteins to also misfold, triggering a chain reaction that causes damage to neural cells. There is no cure for prion diseases and diagnosis is difficult since prion proteins are similar to normal forms.