This document discusses prions, which are infectious agents made solely of protein and responsible for several neurodegenerative diseases such as Creutzfeldt-Jakob disease. It covers the history of prion research, their characteristics, mechanisms of disease, and various forms of transmissible spongiform encephalopathies affecting both humans and animals. Additionally, the document details specific prion diseases, symptoms, and contributing factors to their transmission and pathology.






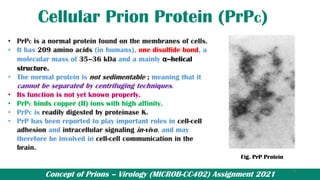

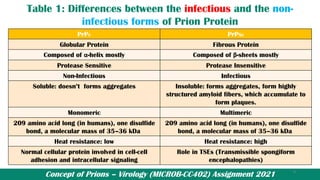



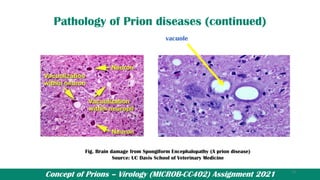






















































![Prions_Presentation[1] [Autosaved]..pptx](https://cdn.slidesharecdn.com/ss_thumbnails/prionspresentation1autosaved-250425151925-c892db5b-thumbnail.jpg?width=640&height=640&fit=bounds)

























