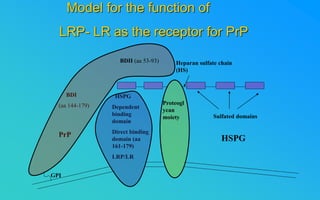
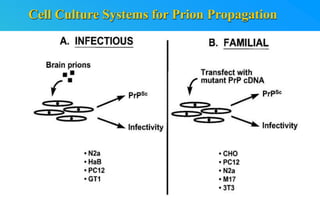
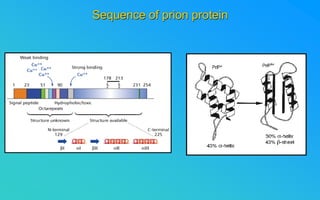
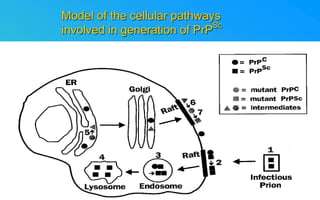
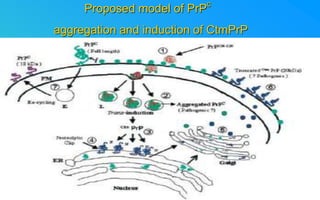
This document discusses prion diseases, which are infectious neurological disorders caused by misfolded prion proteins. It summarizes that prion diseases include Kuru, Creutzfeldt-Jakob disease, and mad cow disease. Prion diseases can be infectious, inherited, or sporadic in origin. The document outlines the differences between normal and misfolded prion proteins and reviews the mechanisms by which misfolded prions are able to induce normal prions to also misfold. It examines the cellular pathways involved in prion propagation and discusses factors that can prevent prion replication.