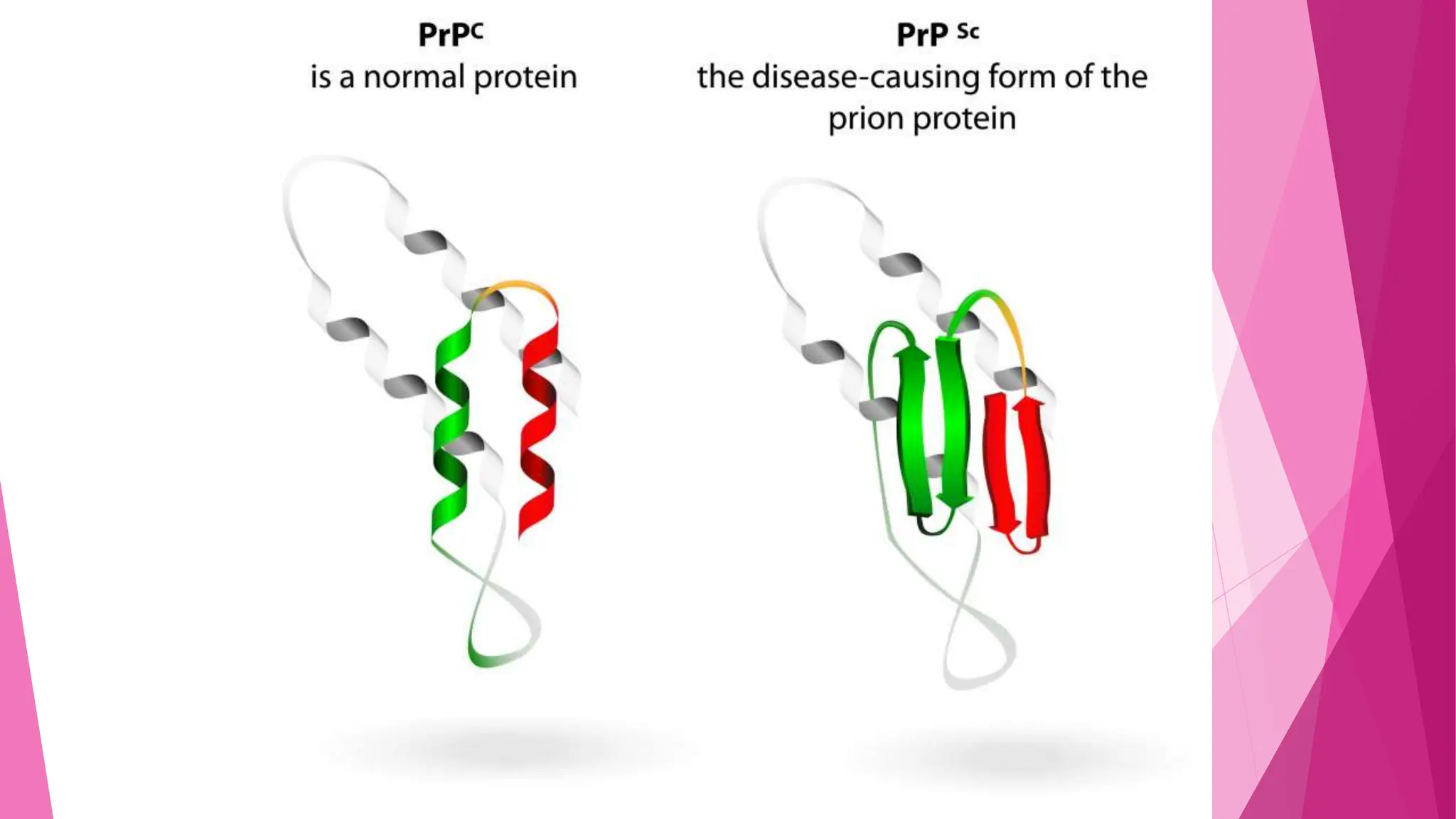
Prion diseases are rare, transmissible brain disorders in humans and animals caused by abnormal prion proteins that lead to neurodegenerative conditions. The most common human forms are Creutzfeldt-Jakob disease (CJD) and variant Creutzfeldt-Jakob disease (vCJD), both of which have no cure and are often diagnosed post-mortem. Prevention includes ensuring proper sterilization of medical equipment and careful consumption of meat from animals potentially affected by prion diseases.