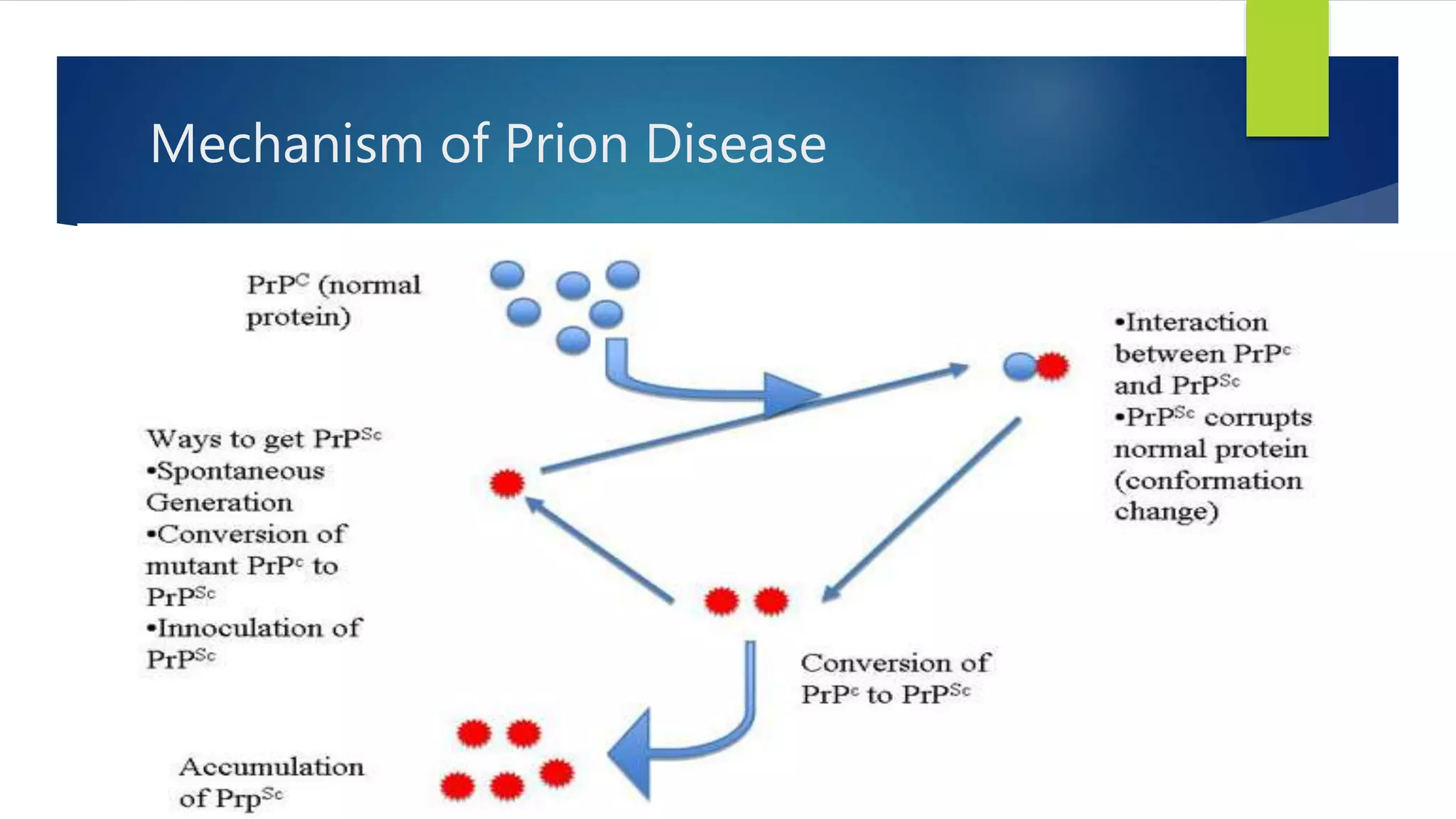
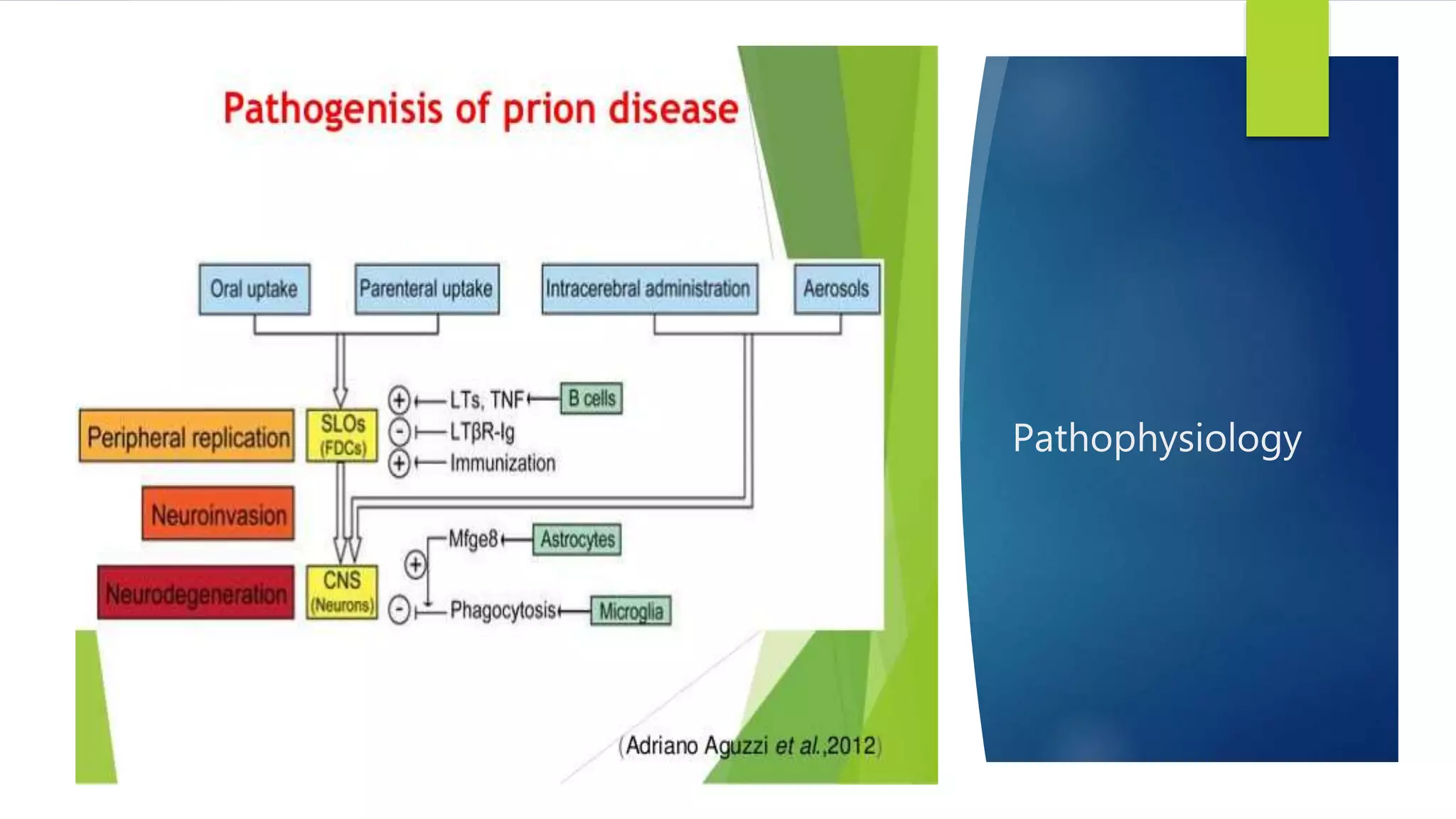
Prion diseases, also known as transmissible spongiform encephalopathies, are rare progressive neurodegenerative disorders caused by prions. Prion diseases affect both humans and animals. There are several types of human prion diseases including Creutzfeldt-Jakob disease, variant Creutzfeldt-Jakob disease, kuru, and Gerstmann-Straussler-Scheinker syndrome. Prion diseases are caused by abnormal folding of the prion protein which forms plaques that damage the brain. There are sporadic, genetic, and acquired forms of prion diseases. Currently, there are no effective treatments for human prion diseases which are universally fatal.