Downloaded 443 times

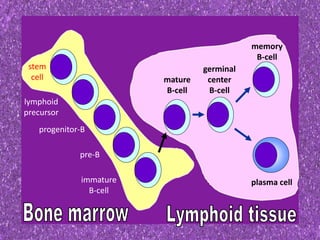
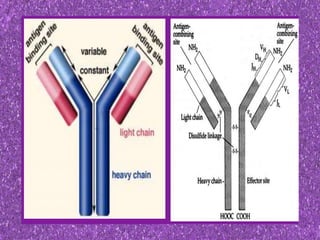
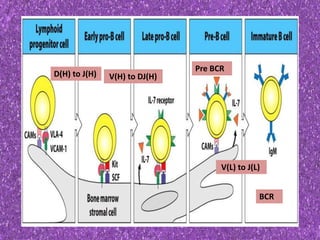
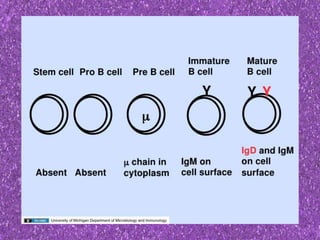



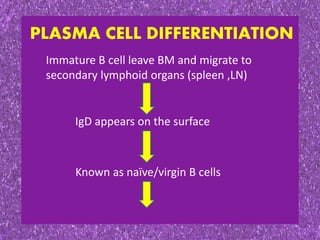
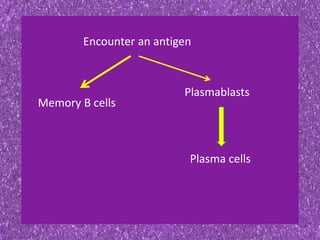
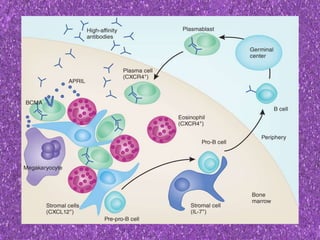
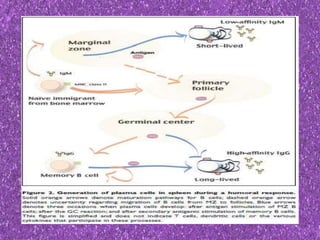




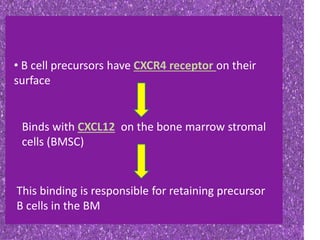

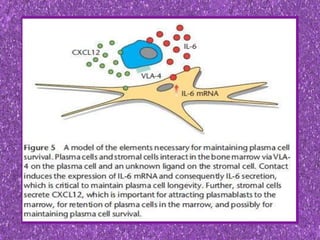
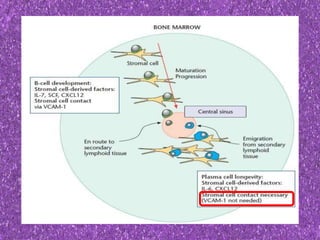


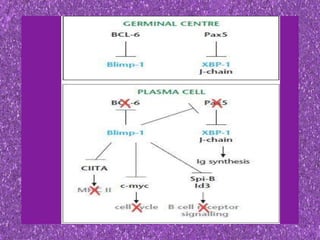


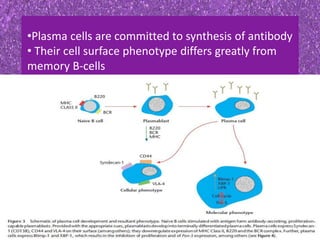

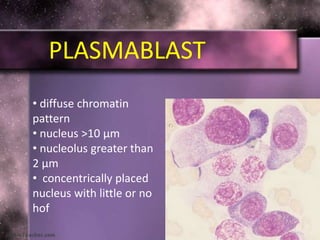
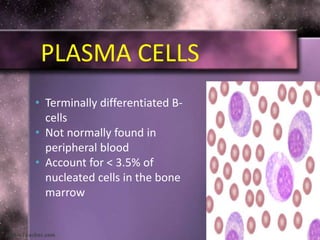
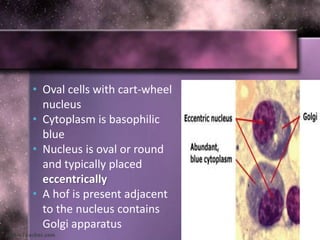
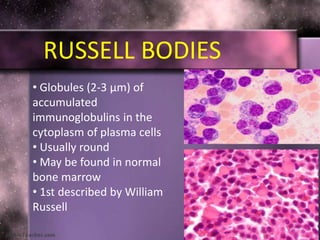










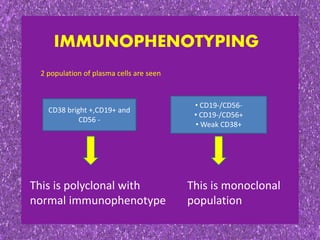



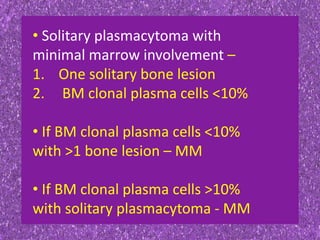


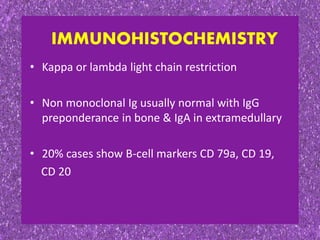






















This document summarizes plasma cell dyscrasias and B-cell development. It discusses the normal development of B-cells from stem cells to plasma cells. It then describes various plasma cell disorders including monoclonal gammopathy of undetermined significance (MGUS), plasma cell myeloma, plasmacytoma, immunoglobulin deposition diseases, and other immunoglobulin secreting neoplasms. It provides details on the morphology, immunophenotyping, classification and diagnostic criteria for these various plasma cell dysrasias.

































































































