Downloaded 99 times



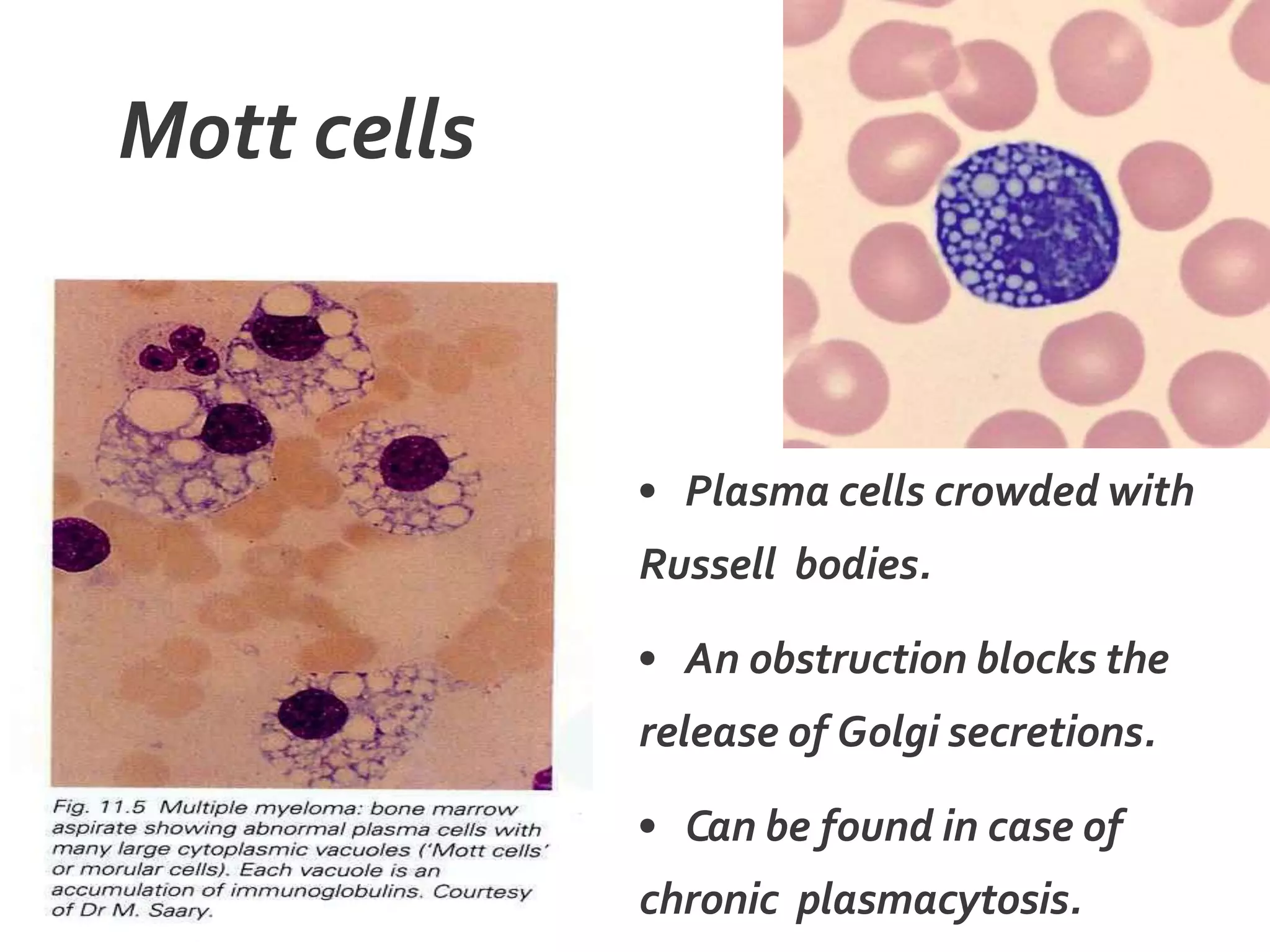
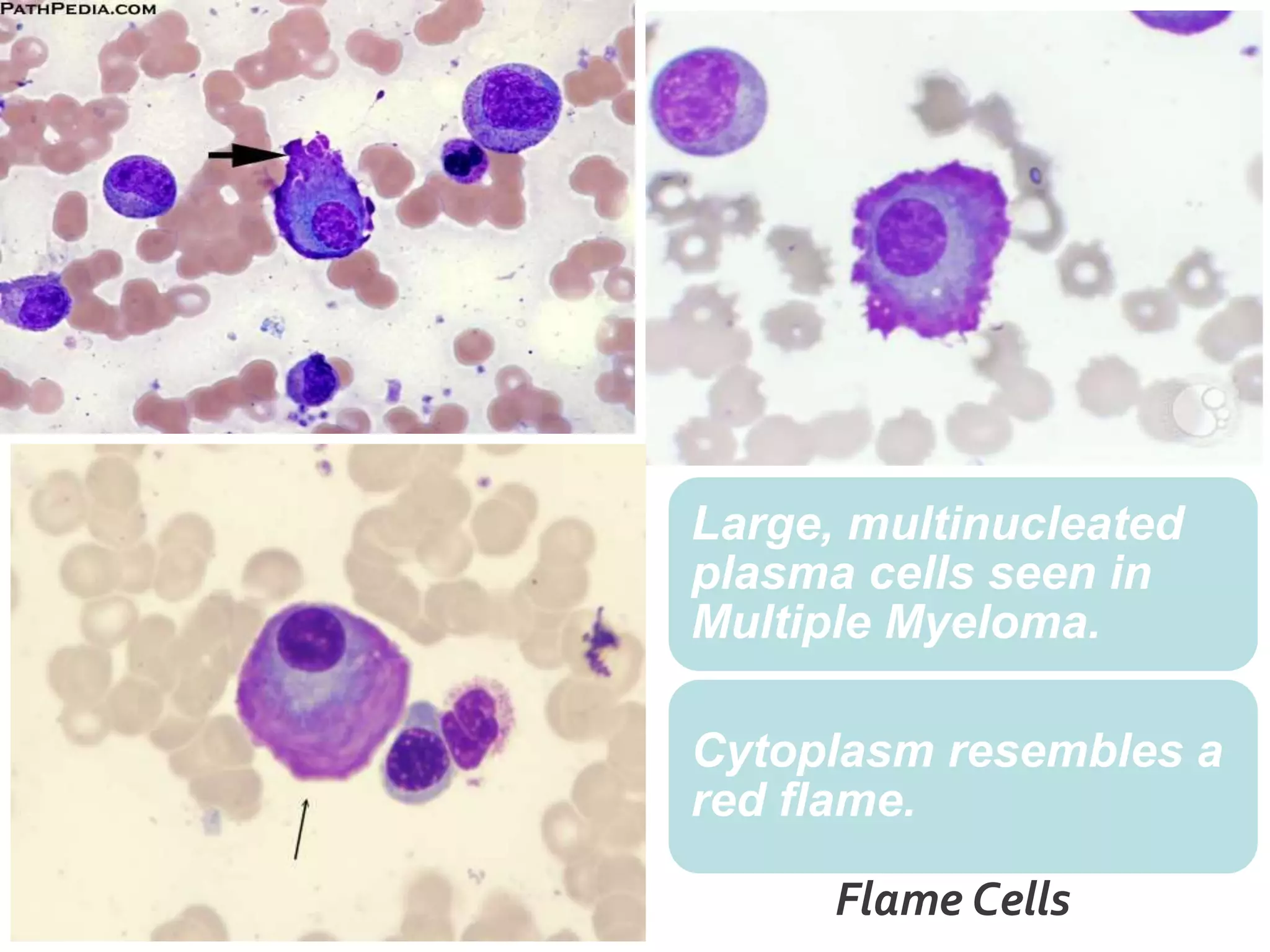



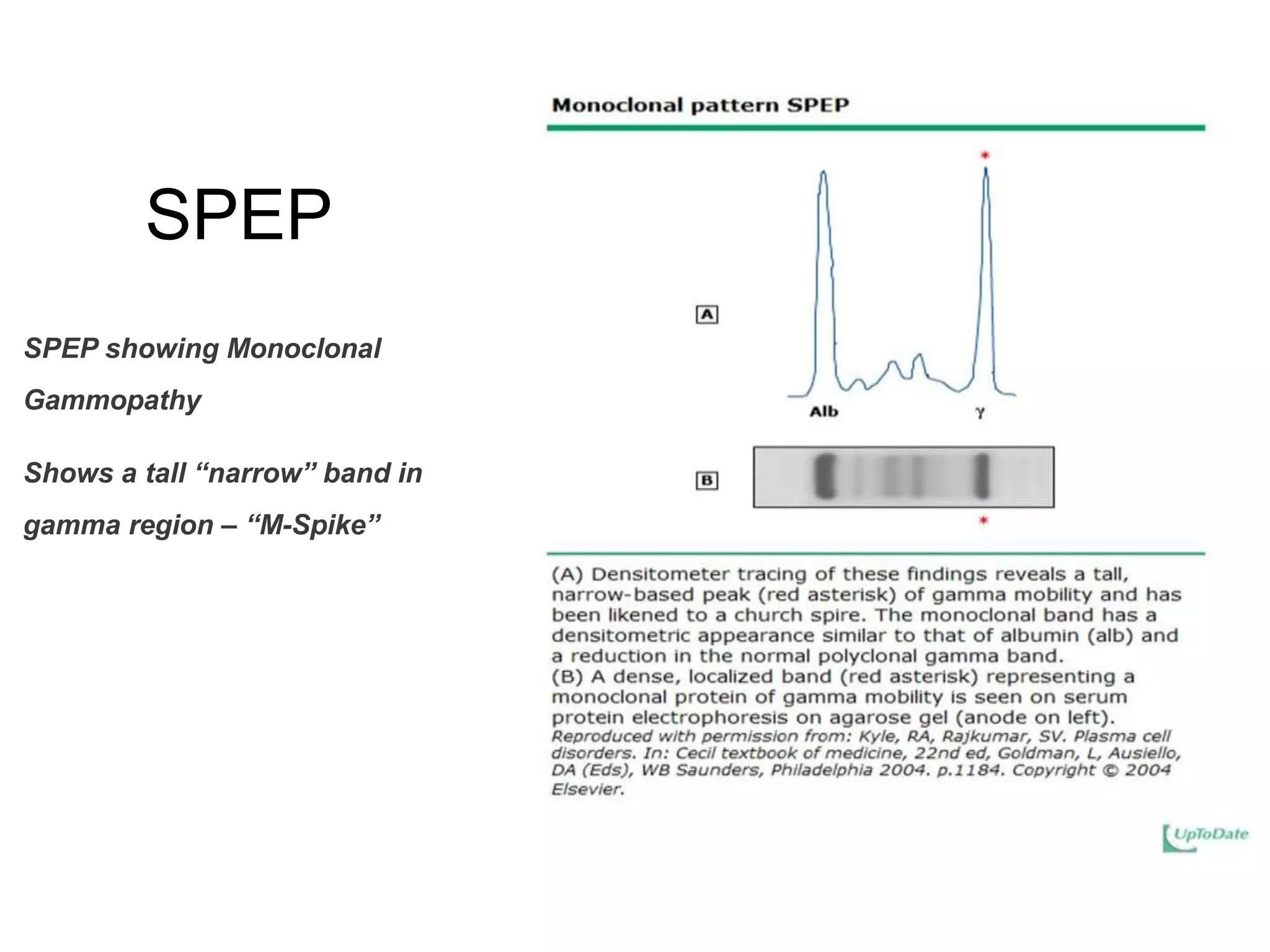
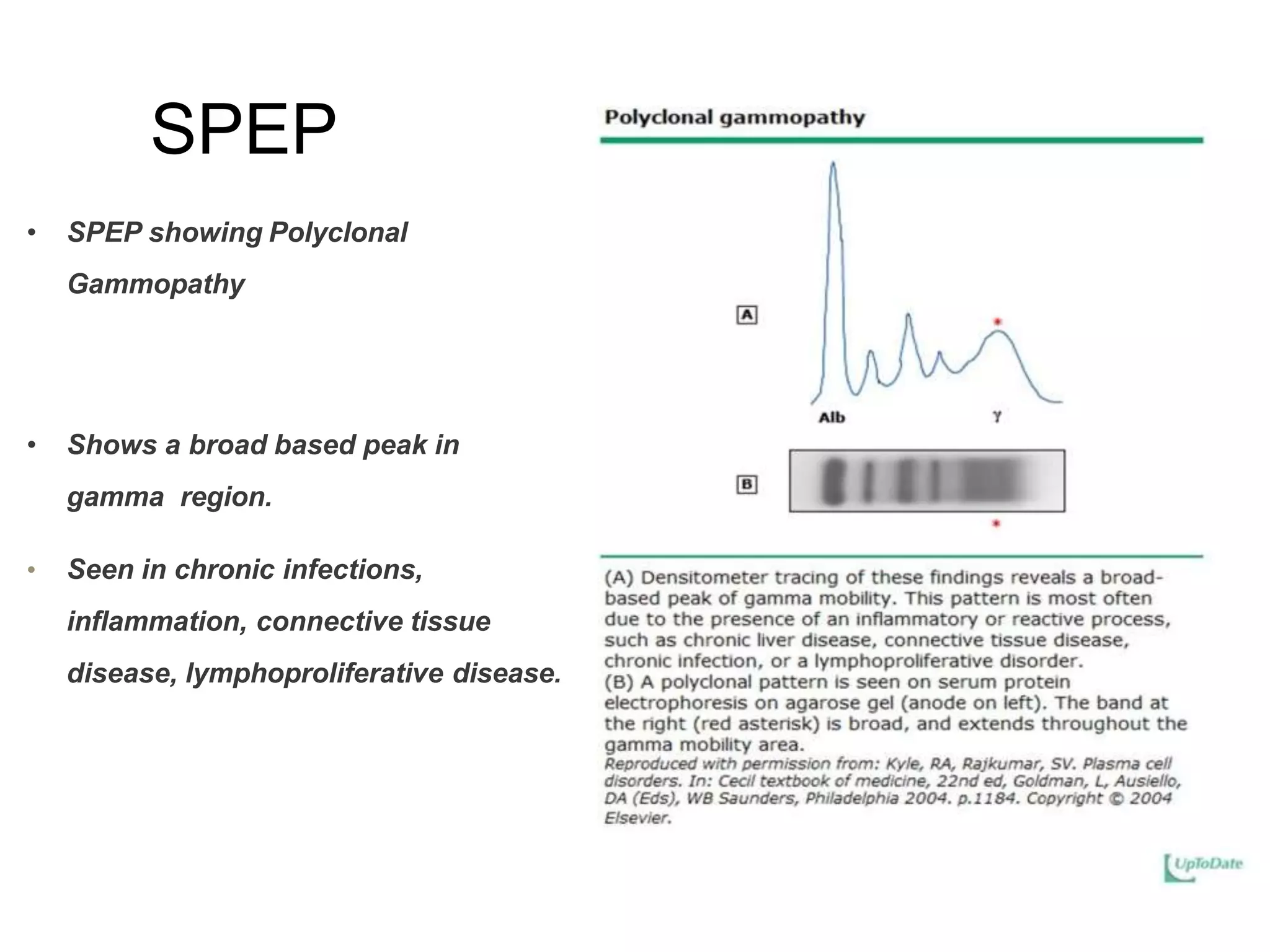

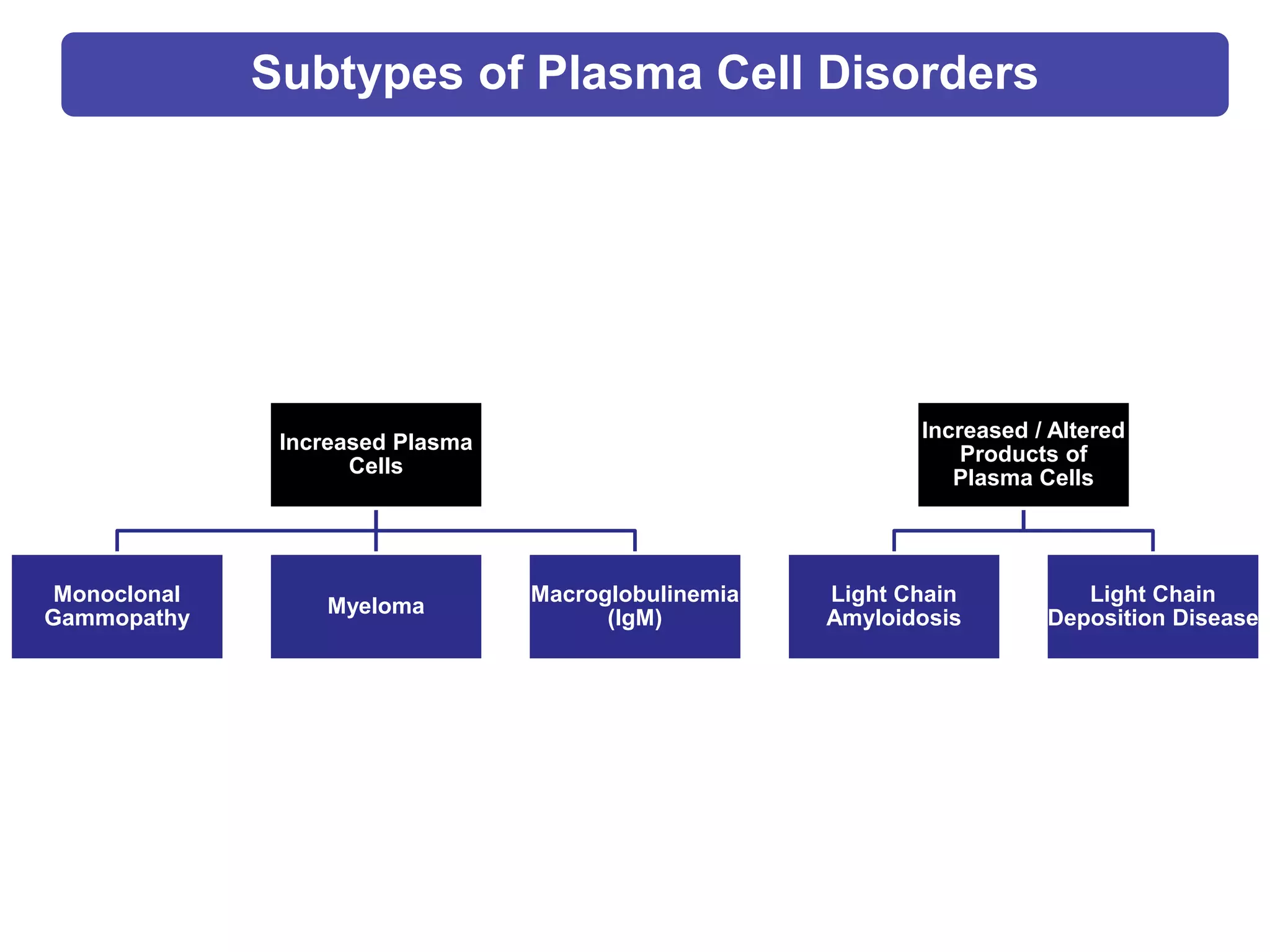


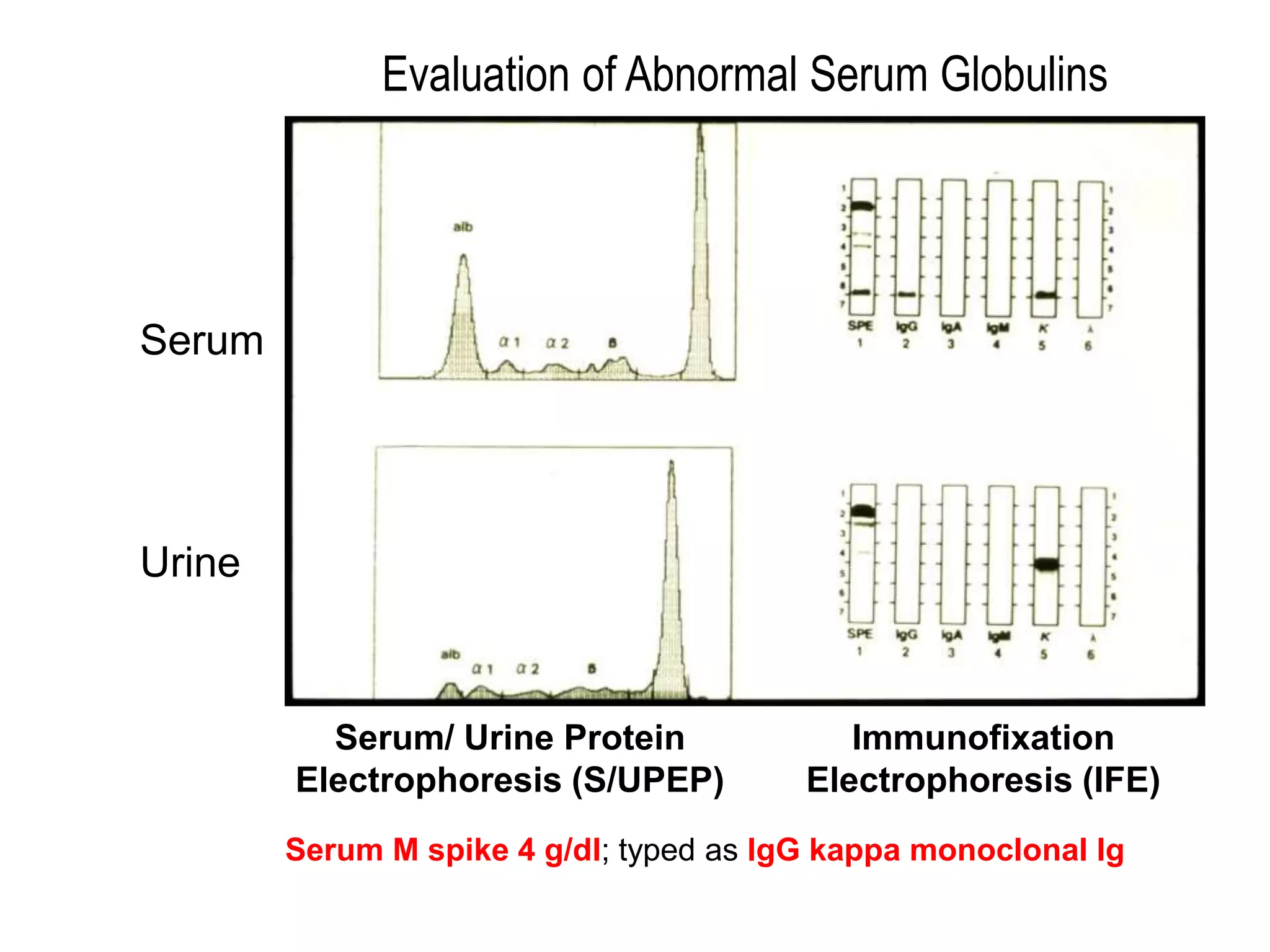


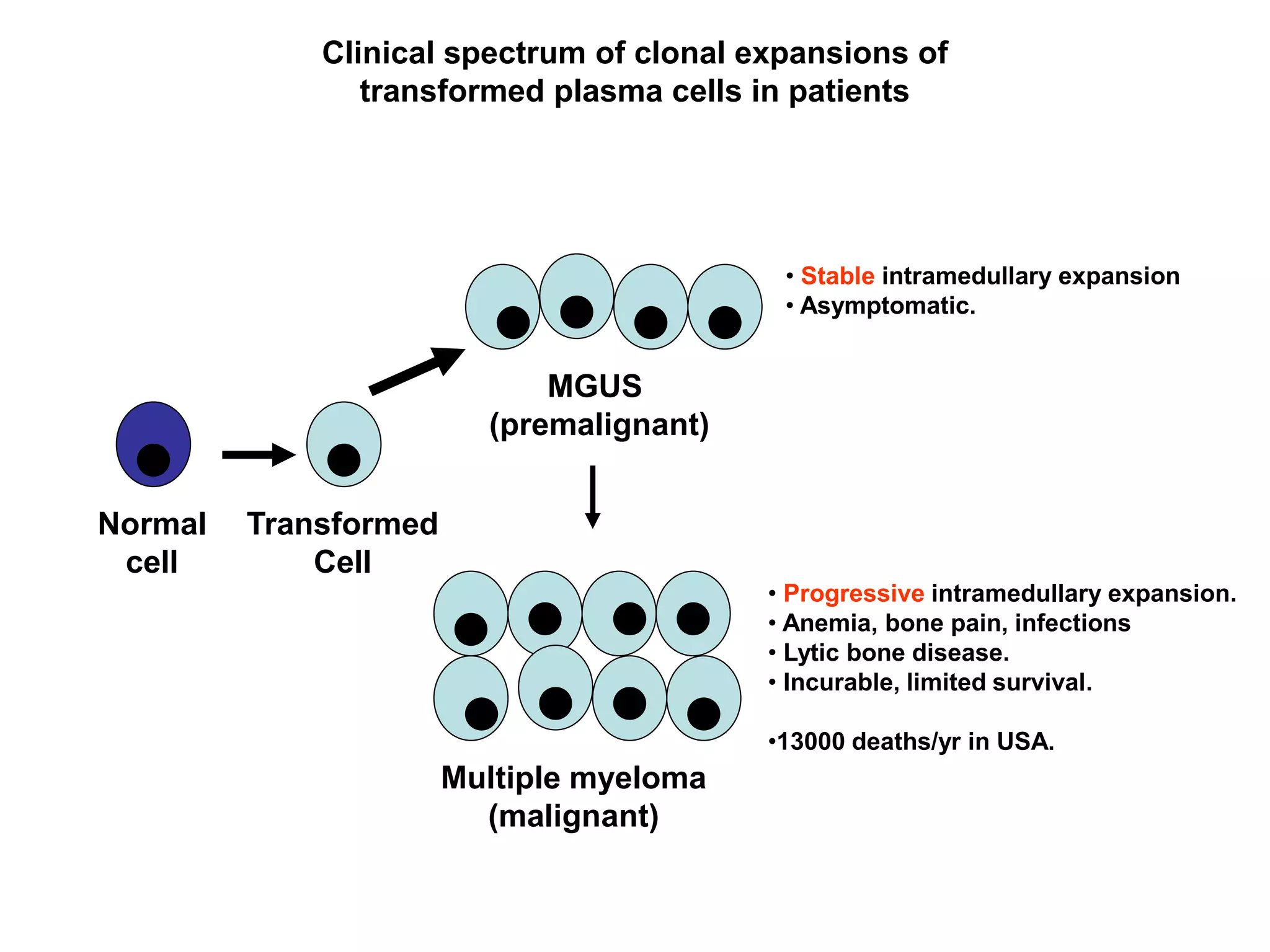


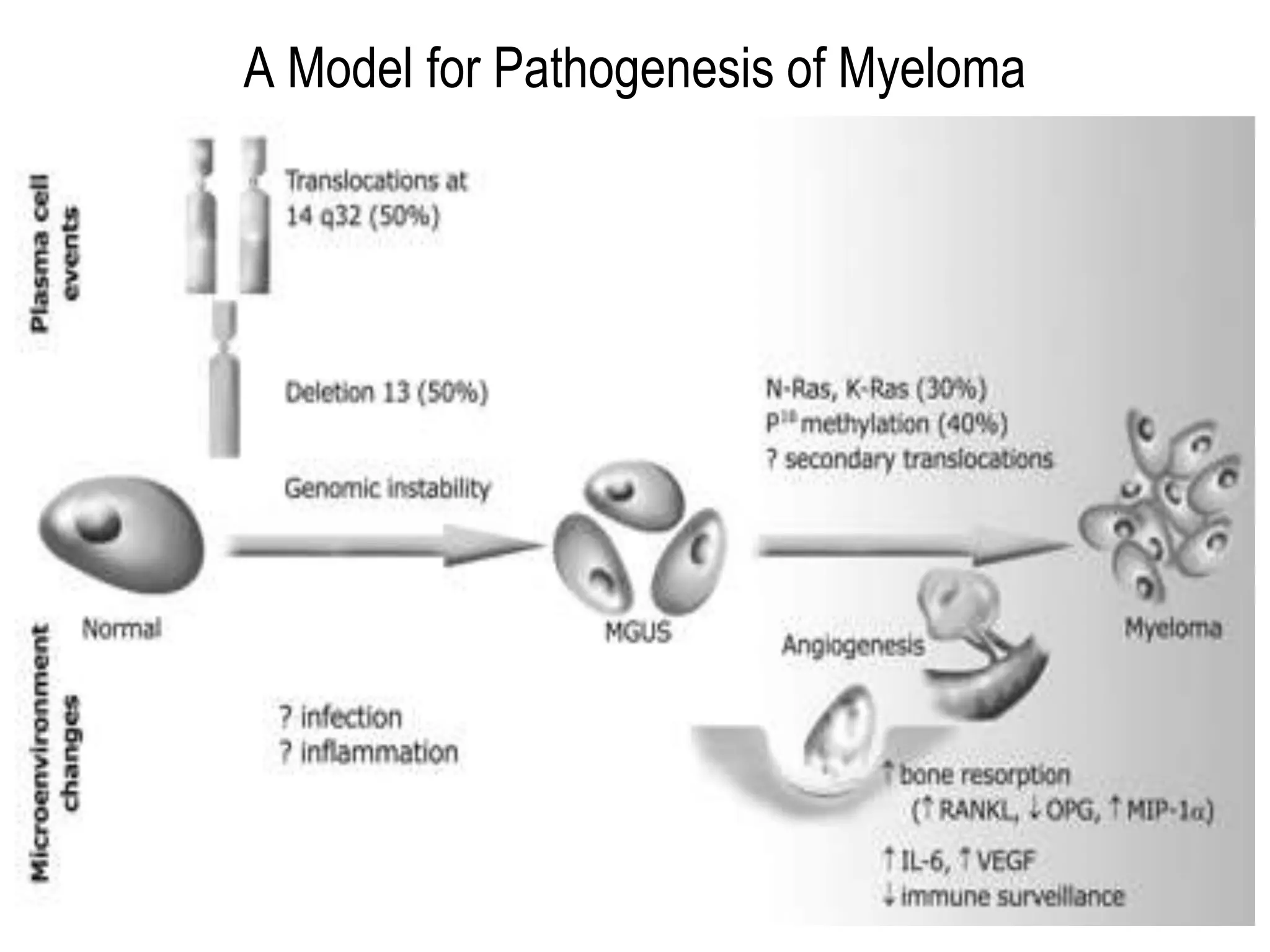



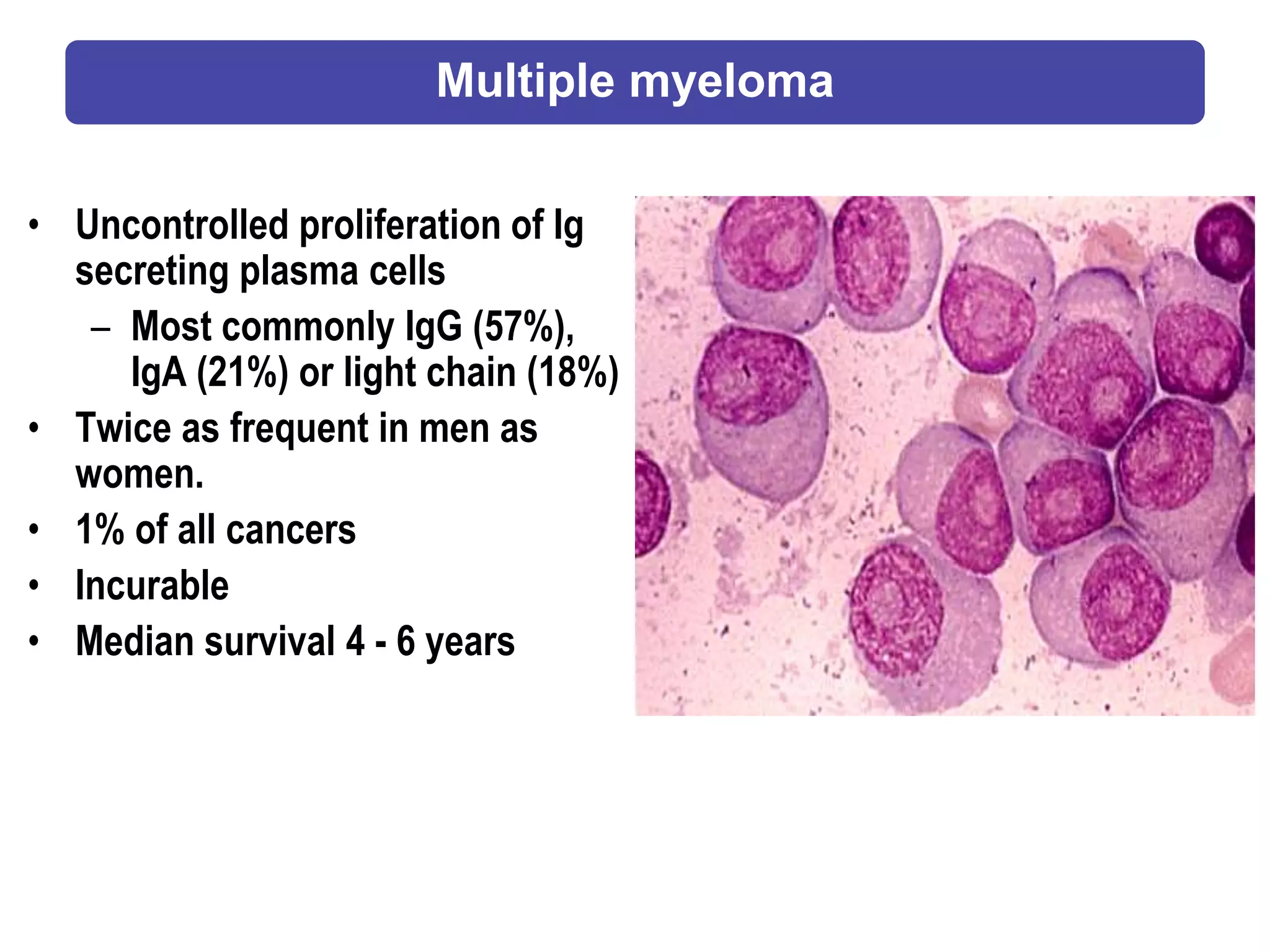


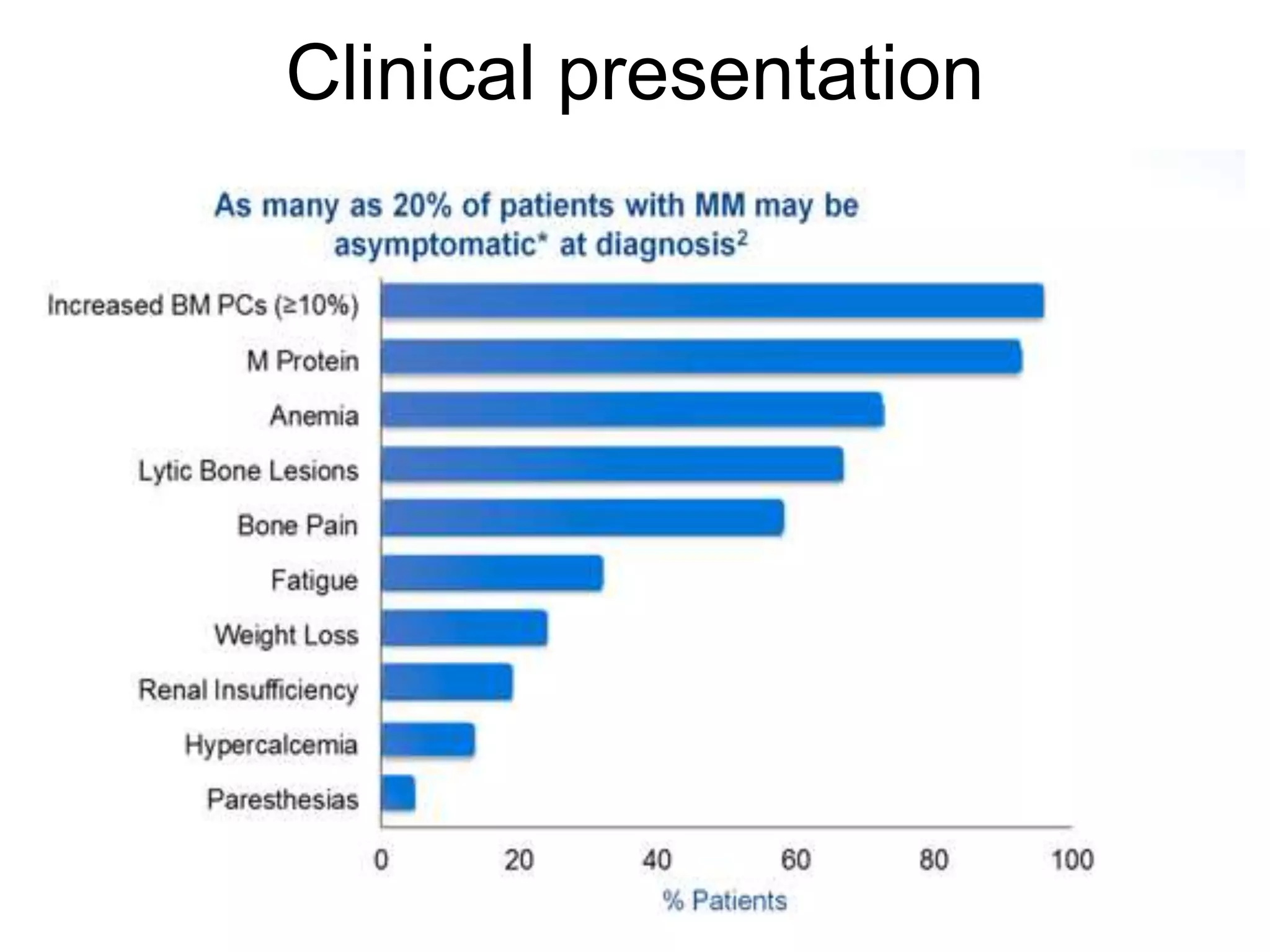
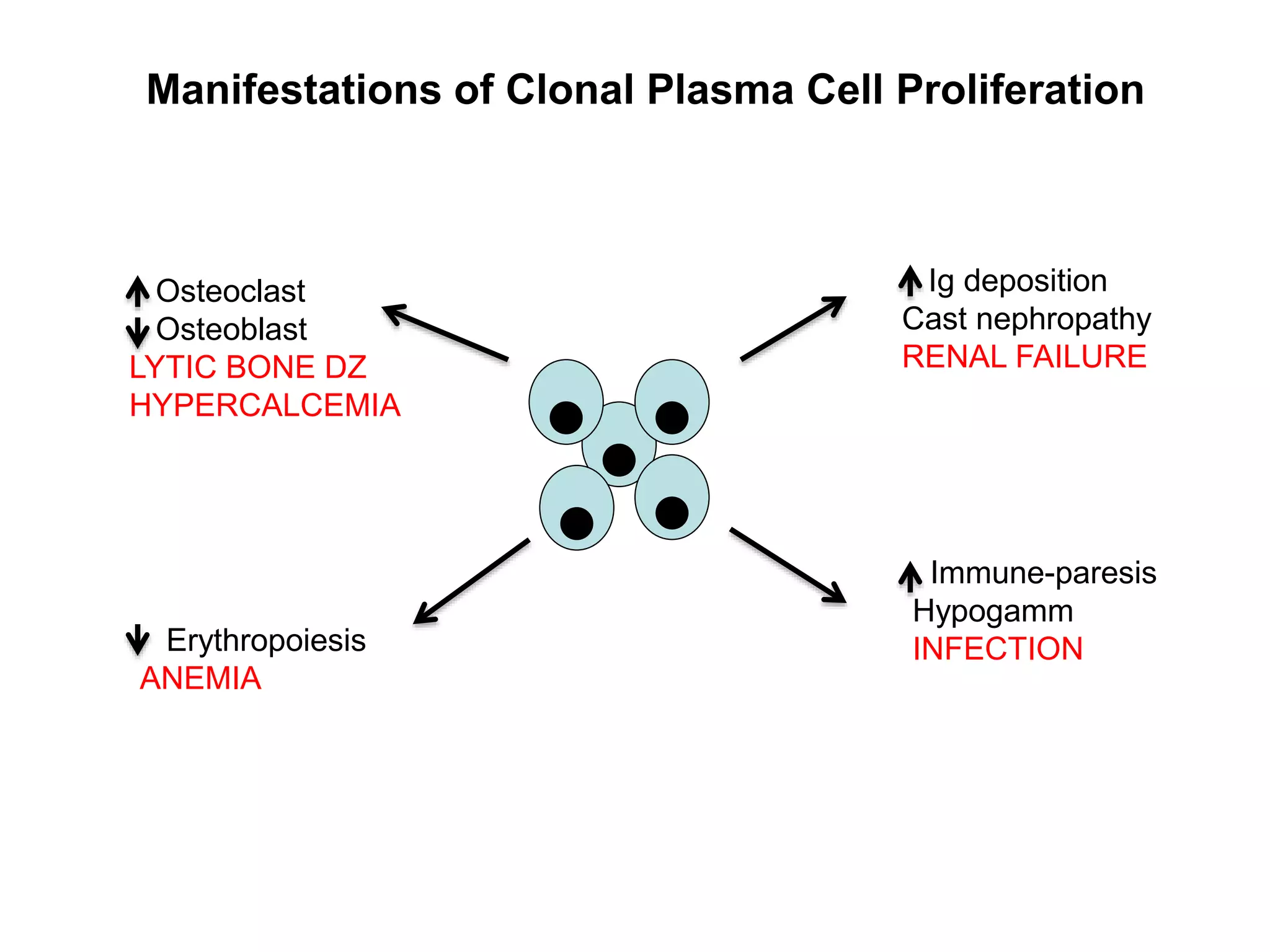




















Plasma cell disorders are a group of lymphoid neoplasms involving the expansion of a single plasma cell clone secreting monoclonal immunoglobulins. Multiple myeloma is a malignant plasma cell disorder characterized by proliferation of plasma cells in the bone marrow, resulting in anemia, bone lesions, hypercalcemia and renal failure. Treatment involves alkylating agents, glucocorticoids, immunomodulatory drugs and proteasome inhibitors. New targeted therapies and personalized treatment approaches based on disease risk factors are improving outcomes.












































































