Downloaded 42 times




![Monoclonal gammopathy of undetermined significance (MGUS).
Plasma cell myeloma and variants:
- Asymptomatic (Smouldering myeloma).
- Non-secretory myeloma.
- Plasma cell leukemia.
Plasmacytoma:
- Solitary plasmacytoma of bone.
- Extraosseous (extramedullary) plasmacytoma.
Immunoglobulin deposition diseases:
- Primary amyloidosis (Ig light chain amyloidosis).
- Systemic light and heavy chain deposition diseases.
Osteosclerotic myeloma : POEMS syndrome:
Plasma cell neoplasms [WHO-2008]](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-5-320.jpg)
![Plasma cell neoplasms [WHO-2017]
Non-IgM monoclonal gammopathy of undetermined significance
Plasma cell myeloma
Plasma cell myeloma variants
- Smouldering (asymptomatic) plasma cell myeloma
- Non-secretory myeloma
- Plasma cell leukemia
Plasmacytoma
- Solitary plasmacytoma of bone
- Extraosseous plasmacytoma
Monoclonal Ig deposition diseases
- Primary amyloidosis
- Light chain and heavy chain deposition diseases [LCDD & HCDD]
Plasma cell neoplasms with associated paraneoplastic syndrome
- POEMS syndrome: polyneuropathy – organomegaly- endocrinopathy – M protein – Skin.
- TEMPI syndrome](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-6-320.jpg)
![Non-IgM
Monoclonal Gammopathy of
Undetermined Significance
[Non-IgM MGUS]](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-7-320.jpg)


![Non-IgM MGUS
Definition:
- Presence of an IgG, IgA, or (rarely) IgD M protein in the serum at a concentration < 3 g/dl
- BM plasma cells < 10%; and
- No end organ damage (CRAB) [hypercalcemia, Renal insufficiency, Anemia, and Bone lesions)
- No amyloidosis attributable to the plasma cell proliferative disorders
Epidemiology:
- Non IgM MGUS= 80% of MGUS.
- Male predominance 60%. [≈ ♂:♀= 2:1]
- Black : White populations = 2:1
Cell of origin: Post-germinal center plasma cells](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-10-320.jpg)
![ Genetic profile:
- Conventional karyotyping are rarely found.
- FISH identifies numerical and/or structural abnormalities in most cases [as myeloma but
prevalence differ].
- Translocations involving the
1. Translocation of IGH locus 50% of cases,
2. t(11;14)(q13;q32) (IGH/CCND1) → 15―25%,
3. t(4;14)(p16.3;q32) (IGH/ NSD2 → 3―9%, and
4. t(14;16)(q32;q23) (IGH/M4F) → 1―5%
5. Hyperdiploidy 40%
6. Deletions of 13q → 35―40%
7. Activating KRAS mutations not detected in MGUS but present in 20% of myelomas](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-11-320.jpg)



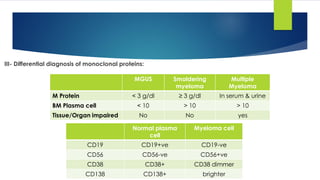
![DD between MGUS [IgM & Non IgM]
Non-IgM MGUS
IgM-MGUS
Plasma cell neoplasm
Lympho-plasma cell lymphoma
Category
85% of MGUS
Male predominance
Black : White = 2:1
15% of MGUS
Male predominance
White > black
Incidence
Post germinal center plasma cell with IgHV gene but
without class switching
B cell with somatic hypermutation
Origin
PB:
- M protein unexpected in PE
- Imm.Fix: IgG 60%, IgA 15%, IgE 1%, IgD 1%
- Light chain 20%.
BM:
Hypercellular BM+ Plasma cells < 10%
BP: Serum IgM < 3 gm/dl
BM: lymphoplasmacytic cell < 10%
Lack of diagnostic feature of LPD, LPL, PCM.
Lab findings
2 populations:
Poly clonal normal Plasma cells: CD19+, CD56-.
Mono clonal aberrant Plasma cells: CD19-,56-/CD19-,56+
Clonal B cells: no specific IPT.
CD19+, 20+, CD5-, 10-, 103-
Plasma cells: 56-
I.P.T
- Serum IgM < 3 gm/dl
- Plasma cell < 10%
- No CRAB nor amyloidosis related to PCM
- Serum IgM < 3 gm/dl
- Plasma cell < 10%
- No CRAB, HSM, LN+, No Hyperviscosity
Diagnostic
Criteria](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-15-320.jpg)

![Plasma cell myeloma
Definition:
- Clonal proliferation of Ig secreting heavy chain class switched terminally differentiated
plasma B cells that produce a monoclonal protein in the serum or urine.
Epidemiology:
- Age: > 50 year not before 30 years. [adult disease]
- Sex: males predominance](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-17-320.jpg)



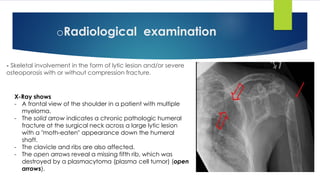
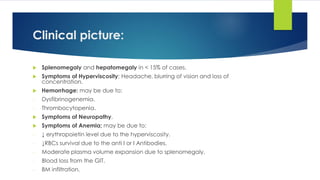
![Clinical picture
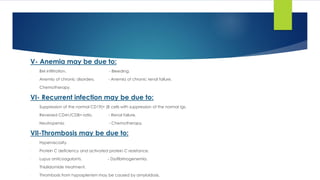
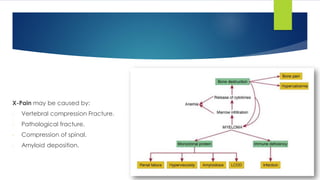
Bone: Back pain, collapse of the vertebrae, osteoporosis and pathological fracture.
Renal disease.
Pain.
Infection.
Hyperviscosity: [Thrombosis tendency] More in IgA due to tendency of IgA to form
polymers.
Amyloidosis: Macroglossia, carpal tunnel syndrome, gastrointestinal disturbance,
neuropathy and liver infiltration.
BM failure:
- Symptoms of anemia: pallor, fatigue, weakness, …..
- Symptoms of neutropenia: recurrent infection.
- Symptoms of thrombocytopenia: bleeding tendency.
CRAB
- Calcium elevation
- Renal Impairment (↑ urea& creatinine)
- Anemia
- Bone (pain, fractures)](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-24-320.jpg)
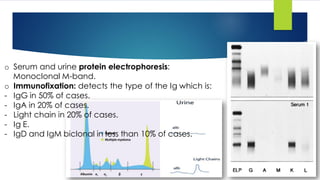
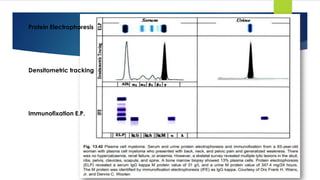
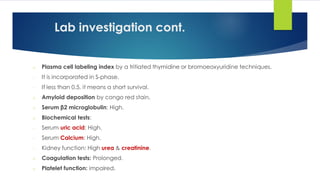
![Laboratory diagnosis
o CBC:
- May be completely normal:
- Anemia.
- Thrombocytopenia & may be Thrombocytosis due to the hyposplenism caused
by amyloid deposition.
o Blood film:
- Blue background due to the high protein.
- Increased reuleaux.
o ESR: Very high>100mm/hr. [Inverted A/G ratio+ hyperviscosity].
o Total protein: High and low serum albumin.
o 24-Hours urine: Bence jones protein.](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-25-320.jpg)

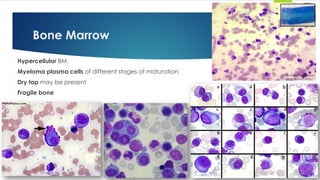

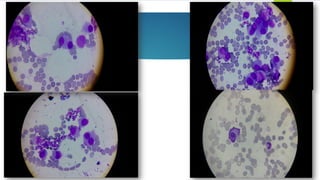
![- Pattern of infiltration: Interstitial clusters or sheets.
- Plasma cells or masses of plasma cells ≥ 30% of BM volume is diagnostic.
- Prominent osteoclastic activity.
- Myelodysaplasia may develop after therapy.
o Immunophenotyping [FCM + IHC]:
- CD19(-), and Surface membrane Ig(-) but it is assigned to B cell lineage because
it is CD79a(+) and cytoplasmic Ig(+).
- Light chain restriction.
- CD38(+), CD138(+), CD56(+) and CD10(+).
- Cyclin D1 is (+) in t(11;14)(q13;32).
Bone marrow biopsy](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-32-320.jpg)
![o Cytogenetic: [Remember 13-14--16-17]
1- Ig heavy chain gene rearrangement of on chromosome (14) with either of the following:
- 11q13 (CCND1).
- 6p21(CCND3).
- 4p16 (FGFR/MMSET).
- 16q23(MAF).
- 20q11(MAFB).
- 8q24(RAS).
2- Dysregulation of cyclin D gene.
3- Micro RNA dysregulation.
4- Aneuploidy:
- Monosomy 13. - (17q-). - (1q -).
- Hyperdiploidy.
Cytogenetic](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-33-320.jpg)





![Smouldering (asymptomatic) myeloma
[No CRAB]](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-43-320.jpg)



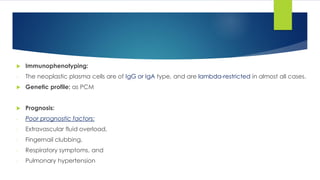
![7- Bone marrow:
- Hypercellular BM with plasma cells less than in MM which may resemble plasma cells or
lymphoplasmacytoid lymphocytes.
8- Immunophenotyping [FCM & IHC]:
- CD19(-), and surface Ig(-) but it is assigned to B cell lineage because it is CD79a(+) and
cytoplasmic Ig(+).
- Light chain restriction.
- CD38(+), CD138(+), CD56(-) and CD10(+).
9- Cytogenetics: t(11;14).
10-Biochemical tests: High serum calcium and creatinine.
Prognosis: very aggressive and resistant to therapy.](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-48-320.jpg)













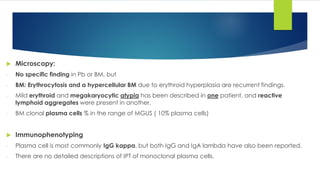
![TEMPI syndrome
Definition: It is a para-neoplastic syndrome associated with a plasma cell neoplasm
It is a rare and recently described disease [11 cases only].
Abbreviation:
- Telangiectasias, Elevated Erythropoietin and Erythrocytosis, Monoclonal gammopathy,
Perinephric fluid collection, and Intrapulmonary shunting.
-
It similar to POEMS syndrome in its manifestations that appear to result from the monoclonal
plasma cell proliferation and associated M protein. However, the clinical and laboratory
findings are mostly distinct from those of POEMS syndrome.
TEMPI syndrome is a rare and only recently described disease.](https://image.slidesharecdn.com/plasmacellneoplasms-new-230123221315-ee4cc8eb/85/Plasma-cell-Neoplasms-2021-pdf-66-320.jpg)




The document provides a comprehensive overview of plasma cell neoplasms, including their classifications such as monoclonal gammopathy of undetermined significance (MGUS), plasma cell myeloma, and variants like smoldering myeloma. It discusses epidemiology, pathogenesis, clinical features, and diagnostic criteria, as well as a focus on genetic abnormalities and laboratory findings associated with these conditions. Additionally, it outlines differential diagnoses and the relationship between various plasma cell disorders and related diseases.























![GRANULOMATOUS LESION OF SKIN2[299] - Read-Only (1).pptx](https://cdn.slidesharecdn.com/ss_thumbnails/granulomatouslesionofskin2299-read-only1-240725163335-974d6cd5-thumbnail.jpg?width=640&height=640&fit=bounds)



































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
















