Downloaded 103 times


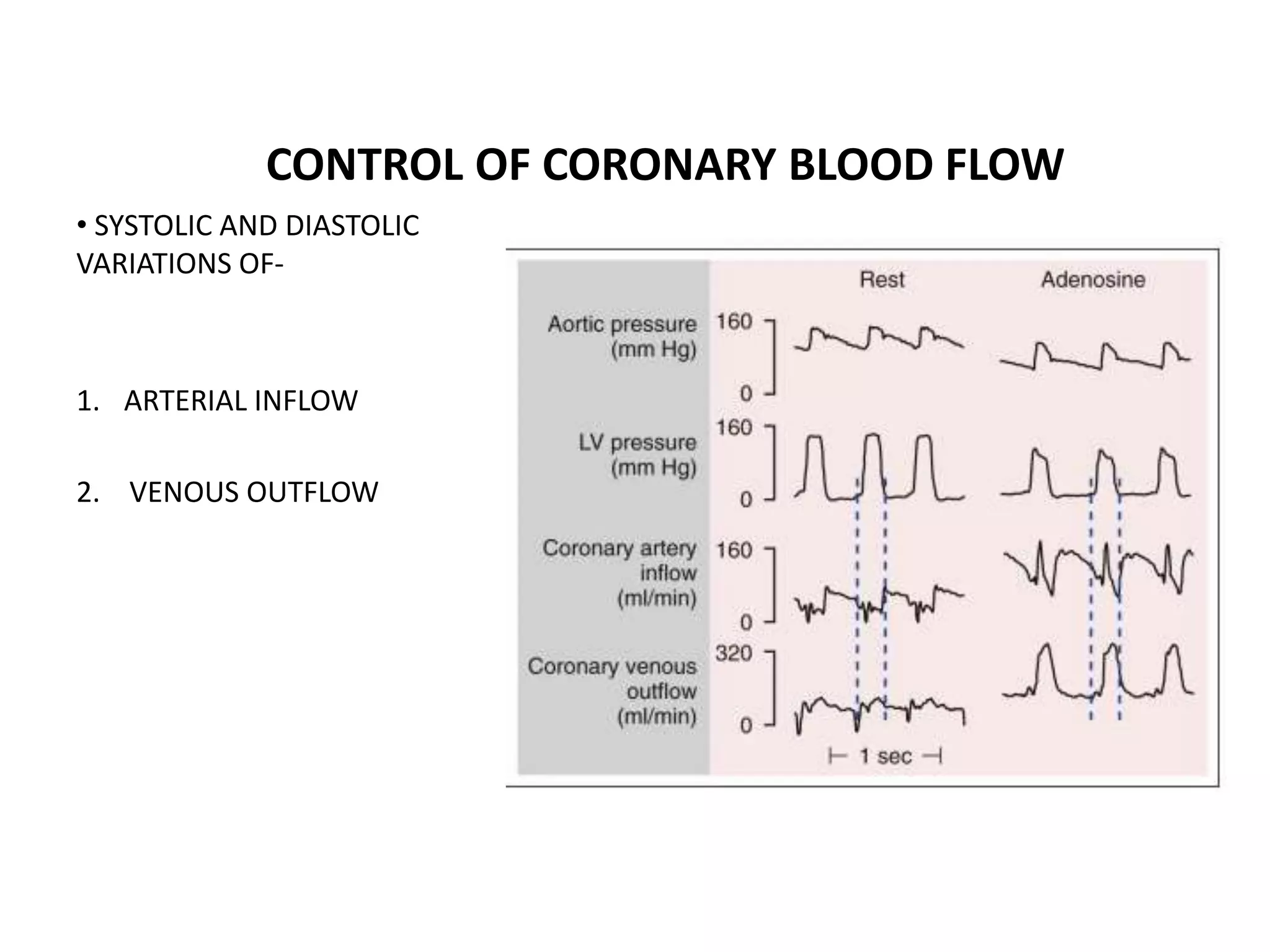



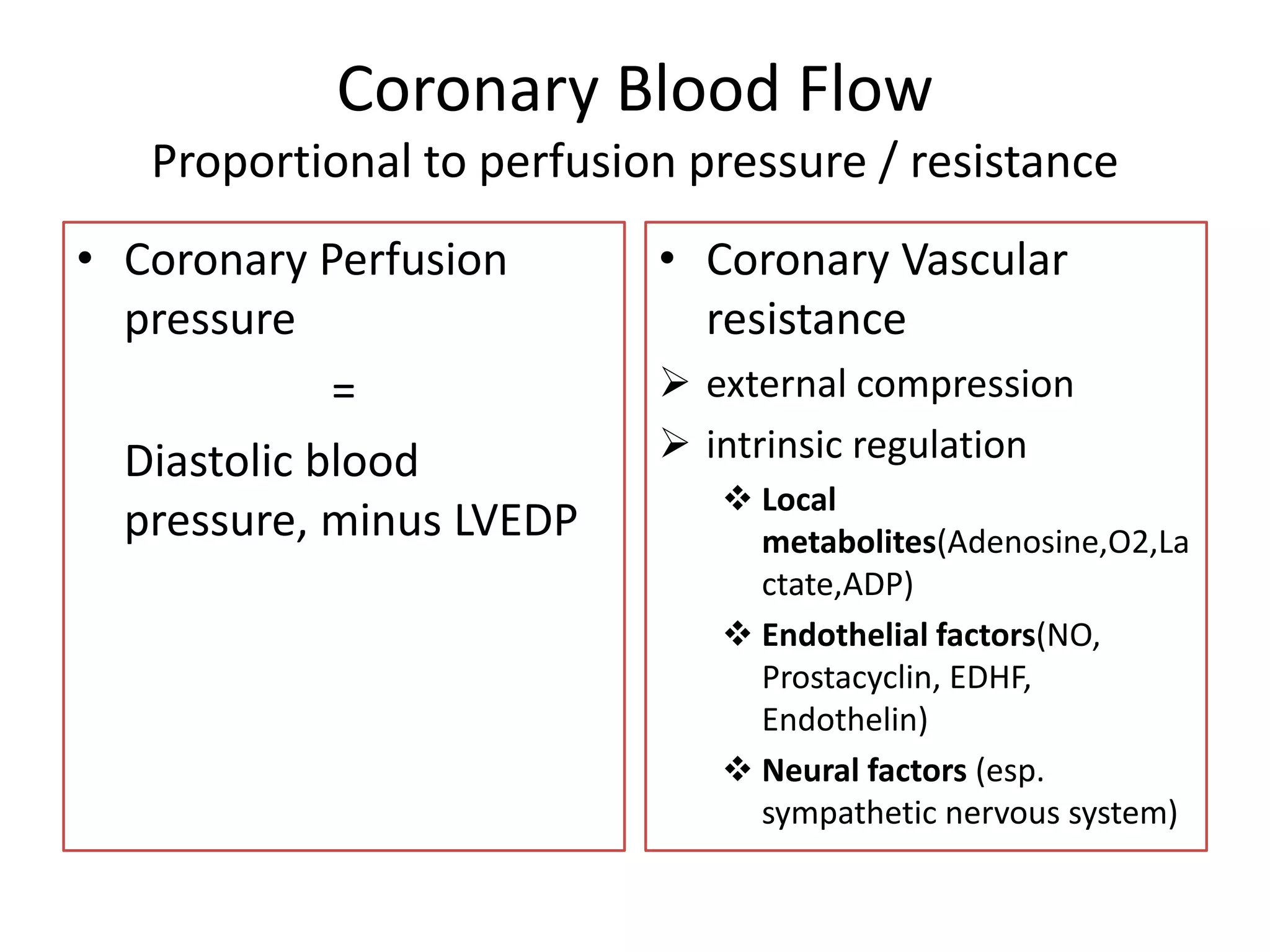






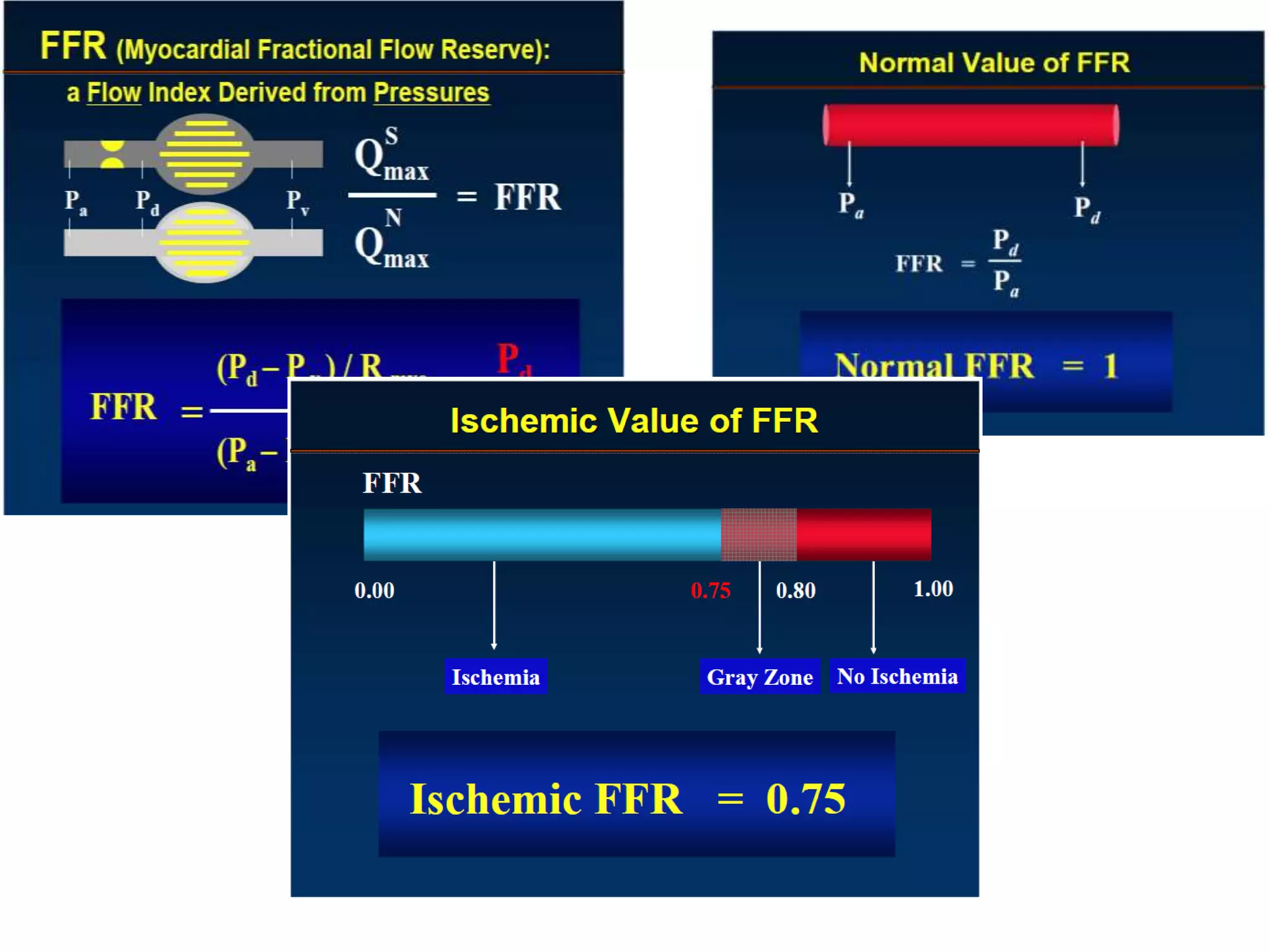










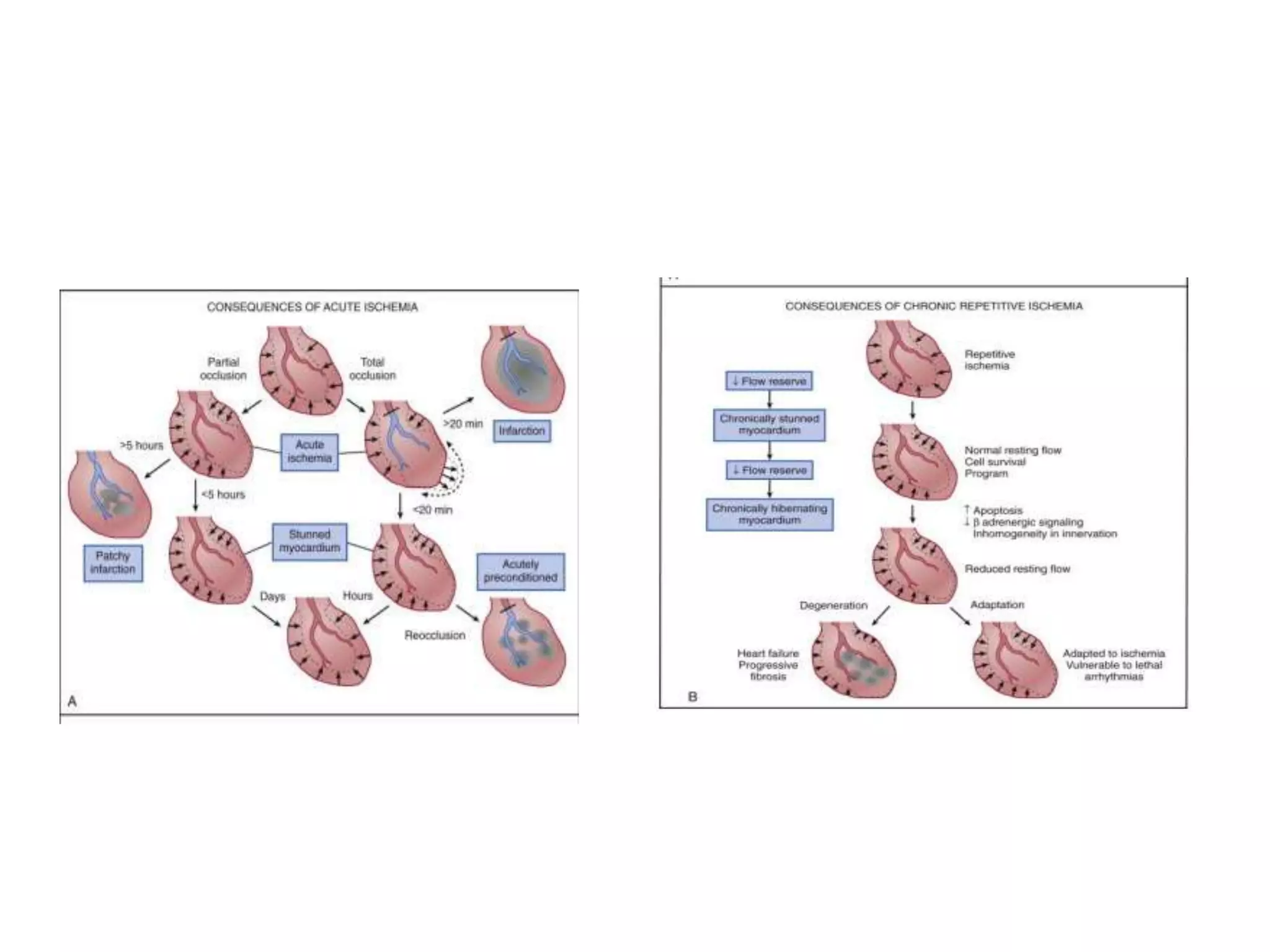








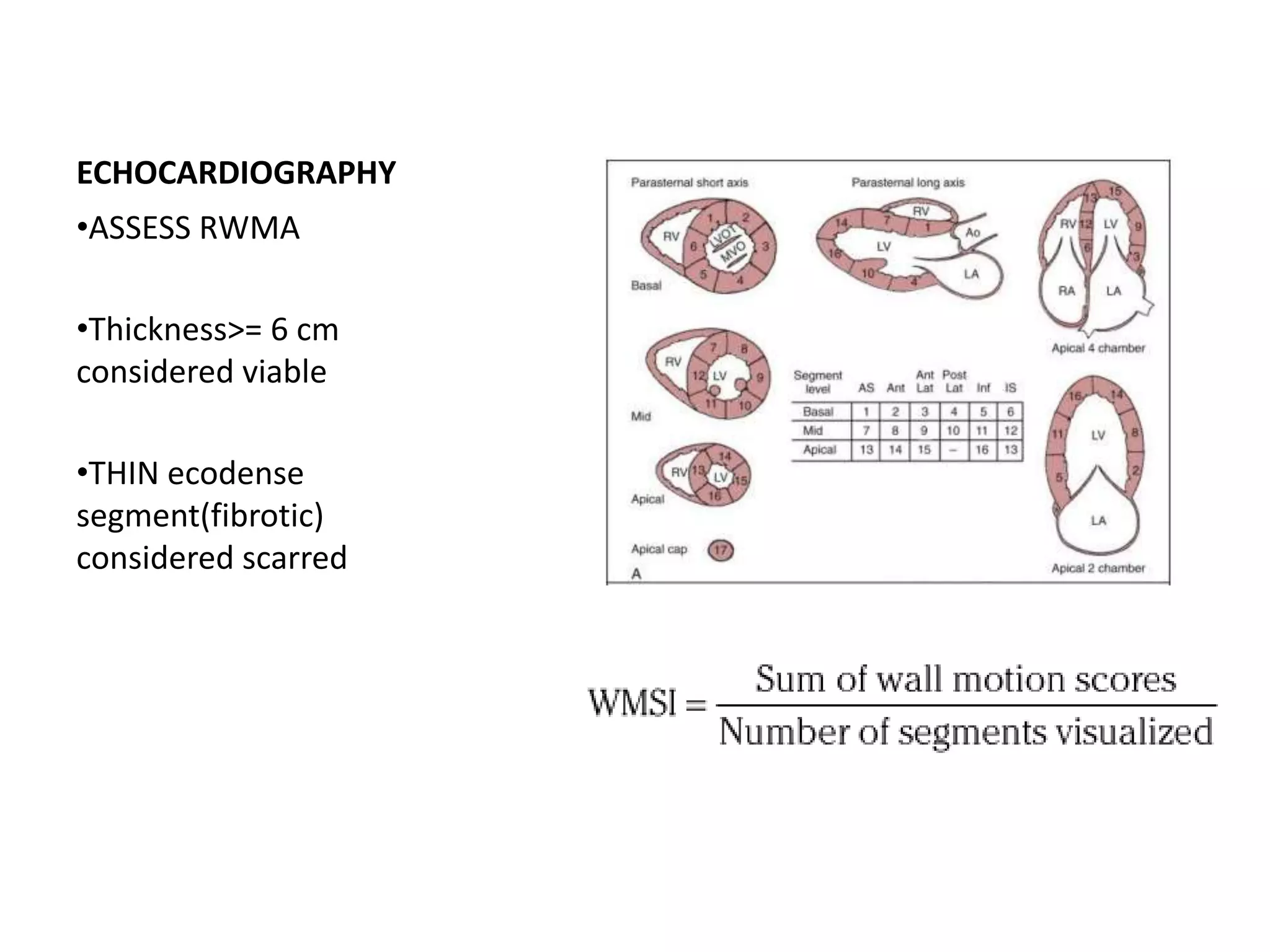
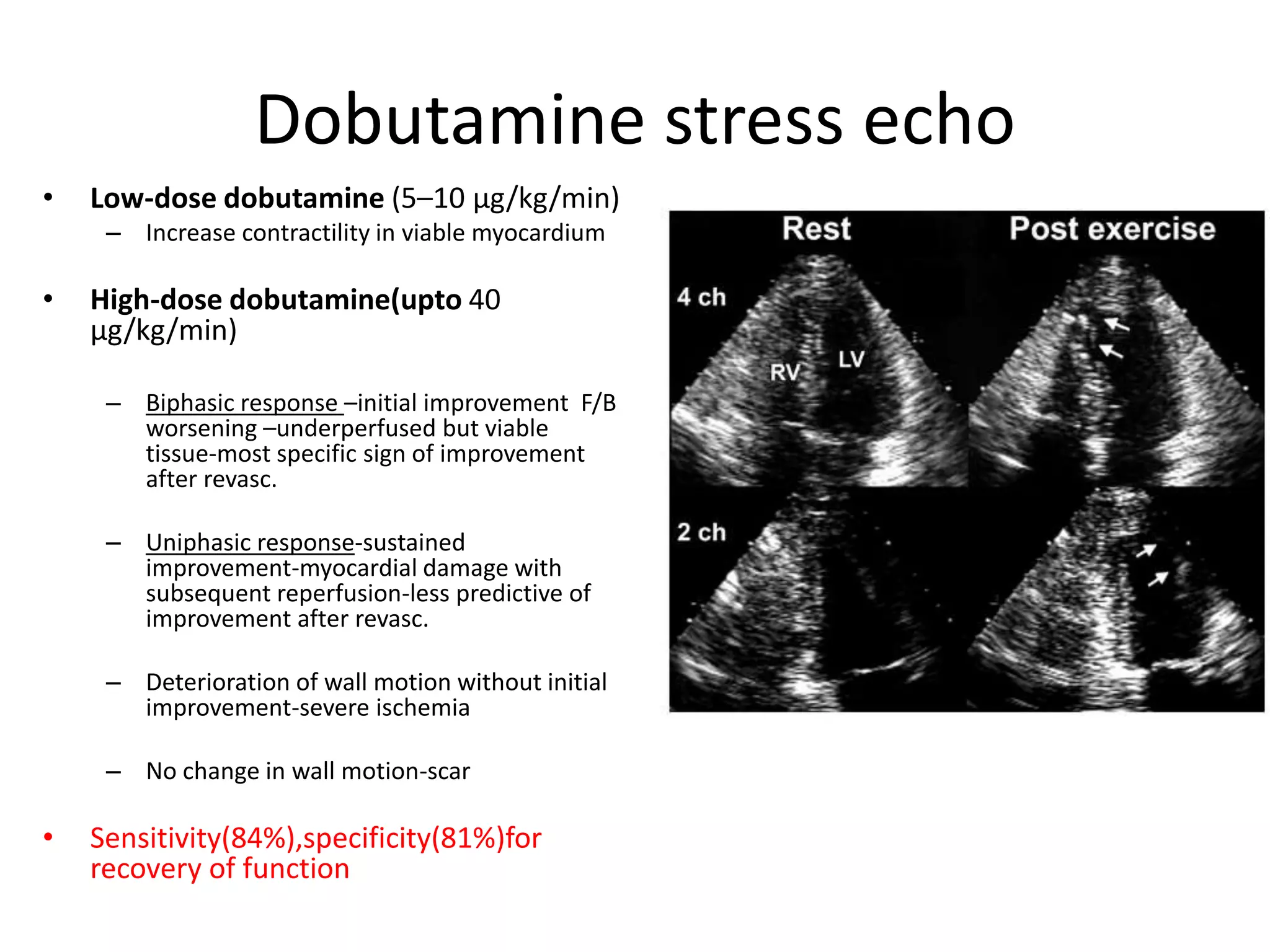










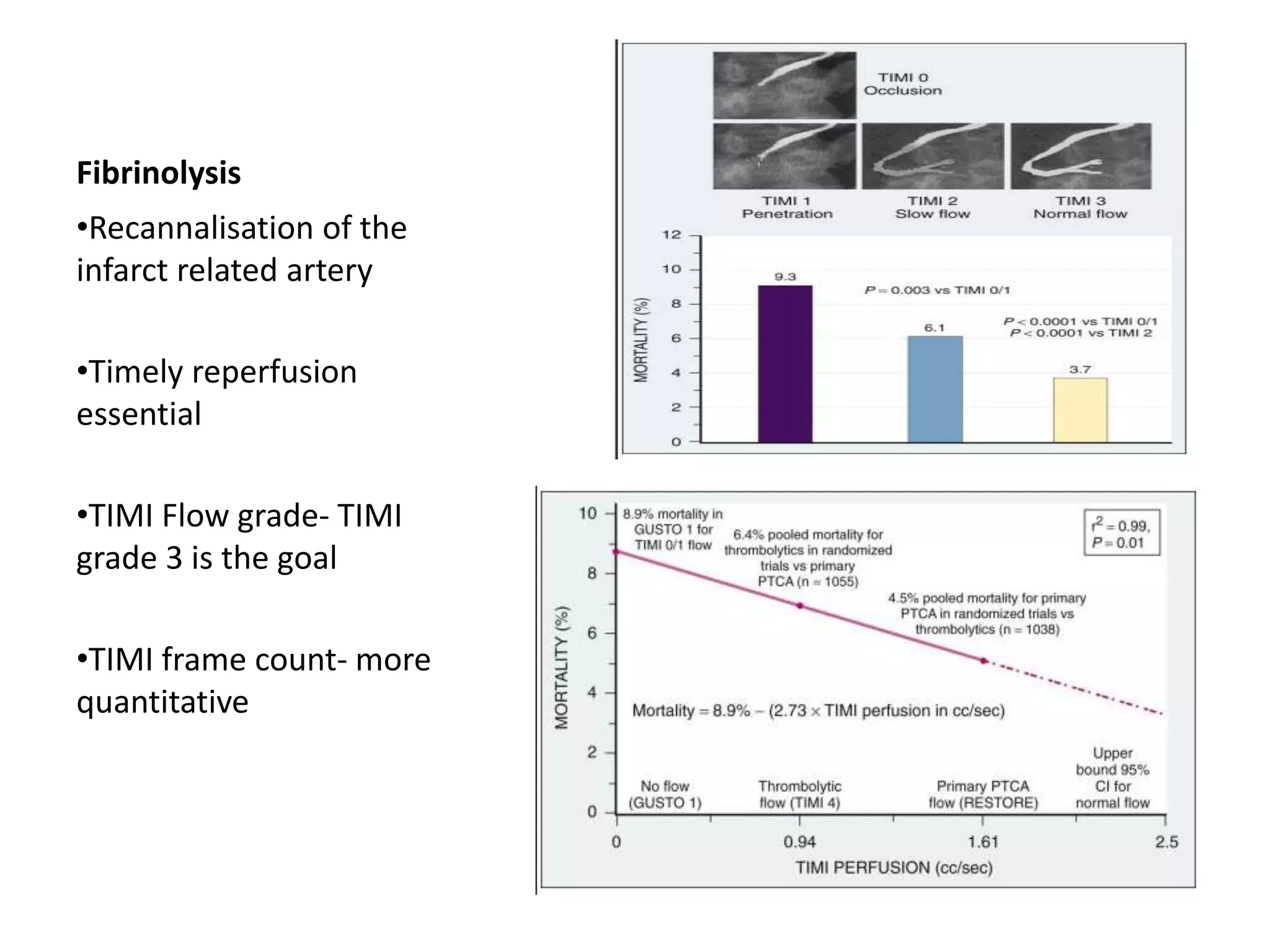

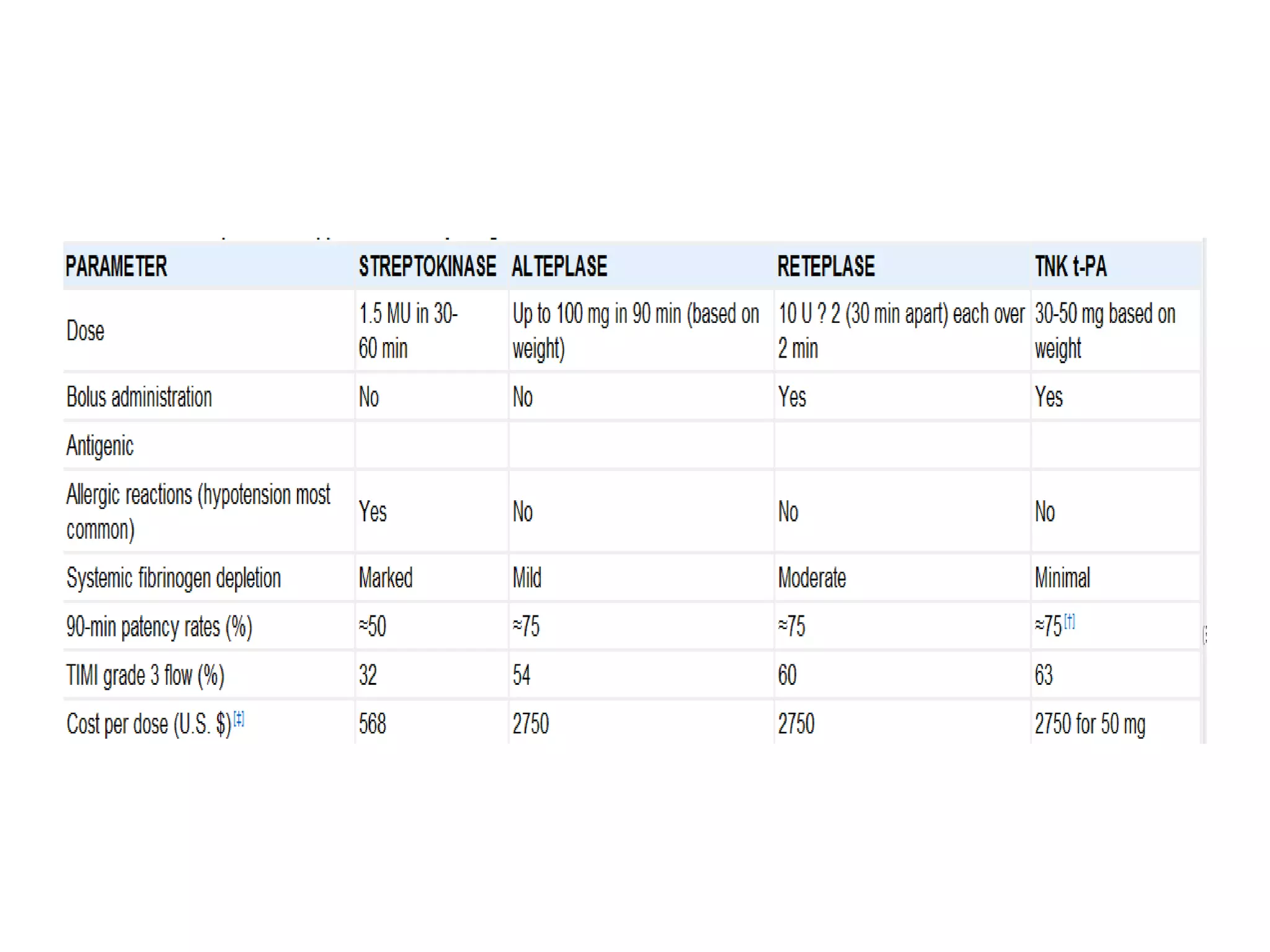







This document discusses the physiological basis of coronary revascularization. It covers topics such as coronary physiology, myocardial viability assessment, and coronary revascularization. Some key points include: - Coronary blood flow is proportional to perfusion pressure over resistance and is regulated by various metabolic and endothelial factors. - Myocardial ischemia occurs when oxygen demand exceeds supply. Coronary autoregulation and flow reserve help maintain adequate flow. - Myocardial viability refers to dysfunctional tissue with limited scarring that has potential for functional recovery after revascularization through mechanisms like stunned myocardium and hibernation. - Various techniques can assess viability including cardiac imaging and evaluating improvement in function after revascularization. Viability assessment aids decisions about revascularization

























































































