Downloaded 27 times


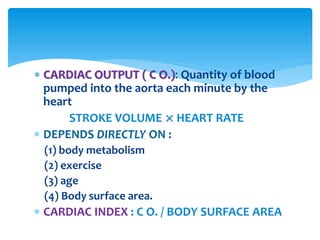
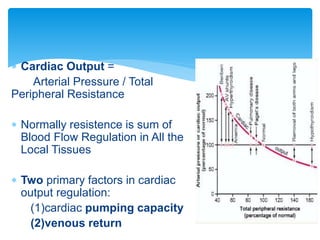



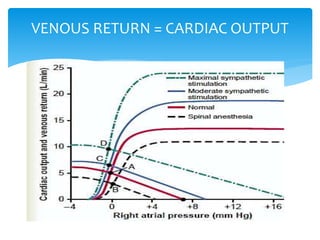





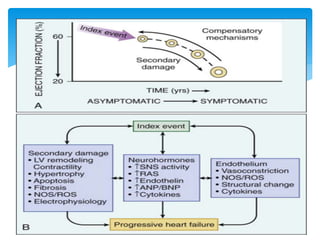


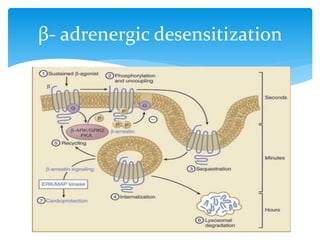











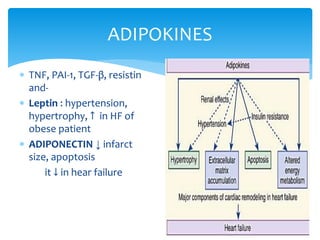


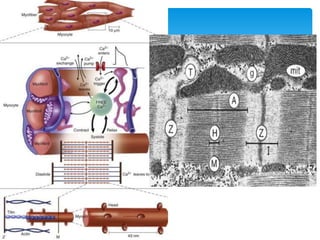








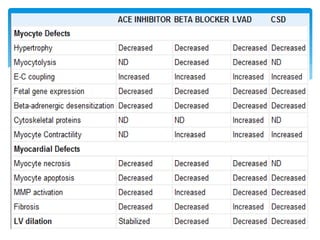





Heart failure occurs when the heart cannot pump enough blood to meet the body's needs due to structural or functional abnormalities. It is characterized by symptoms like dyspnea and fatigue. The pathophysiology of low cardiac output heart failure involves neurohormonal alterations that impair the heart's pumping ability and venous return. The sympathetic nervous system and renin-angiotensin-aldosterone system become overactivated, causing further cardiac remodeling and dysfunction over time. Oxidative stress, inflammation, and other factors also contribute to the progression of heart failure.











































































