



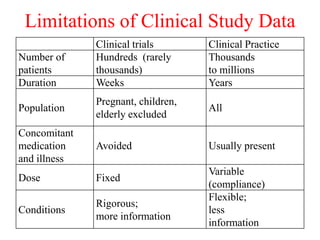
























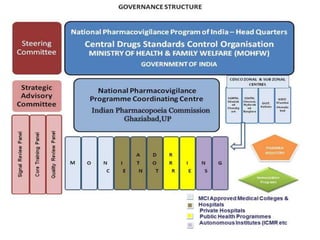



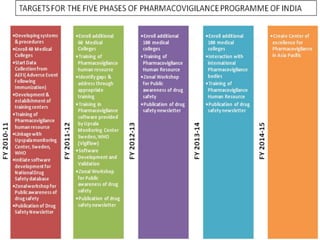
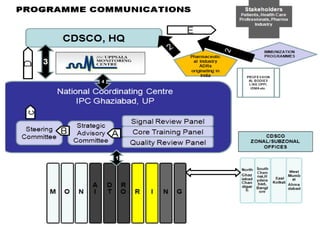


Pharmacovigilance is the science of monitoring approved drugs to detect adverse effects. It aims to improve patient safety by understanding drug risks. Clinical trials cannot detect all risks due to limited size and duration. Spontaneous reporting allows healthcare providers to report suspected adverse drug reactions. Limitations of clinical data and withdrawals like thalidomide led to pharmacovigilance programs worldwide including the WHO program and national programs in India, UK, and US. Pharmacovigilance involves collecting, analyzing, and communicating safety information to improve patient therapy and public health.































































































