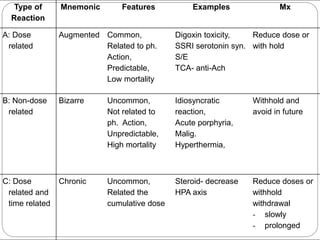
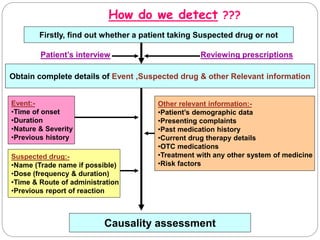
This document discusses pharmacovigilance, which involves monitoring the safety of drugs after they have been approved. It defines pharmacovigilance and explains why it is needed given limitations of clinical trials. It describes types of adverse drug reactions and how they are classified. It outlines the goals and processes of pharmacovigilance programs, including reporting adverse reactions, conducting causality assessments, and submitting periodic safety update reports. The overall aim is to ensure safe and effective use of medicines through continual monitoring and regulatory action.


































































































































![Monoclonal antibodies [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/monoclonalantibodiesautosaved-150310003637-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)



















