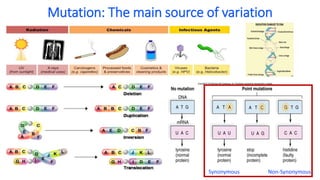
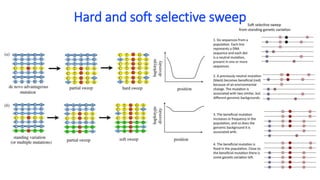
Neutral theory proposes that most mutations are neutral and do not affect fitness. Under neutral evolution, genetic drift is the main factor driving changes in allele frequencies in populations rather than natural selection. Tests based on polymorphism and divergence data can help determine if loci are evolving neutrally or under the influence of selection. Extended haplotype homozygosity (EHH) and cross population EHH (XPEHH) are methods used to detect signatures of positive selection by examining the breakdown of linkage disequilibrium around candidate regions.

































![Measures of Linkage Disequilibrium (LD)
At linkage equilibrium
D = PABPab-PAbPaB = 0
or, PABPab = PAbPaB
At LD PABPab ≠ PAbPaB
If D is negative (more heterozygous gamets),
D’ = D/Dmin, [Dmin = the larger of -pApB and –qaqb]
If D is positive (more homozygous gamets),
D’ = D/Dmax [Dmax = the smaller of pAqB and qapb]
(Correlation between the loci)
(D’= Recombination
dependent measurement)](https://image.slidesharecdn.com/neutraltheory2019-200709035119/85/Neutral-theory-2019-36-320.jpg)















































































