Download as PDF, PPTX



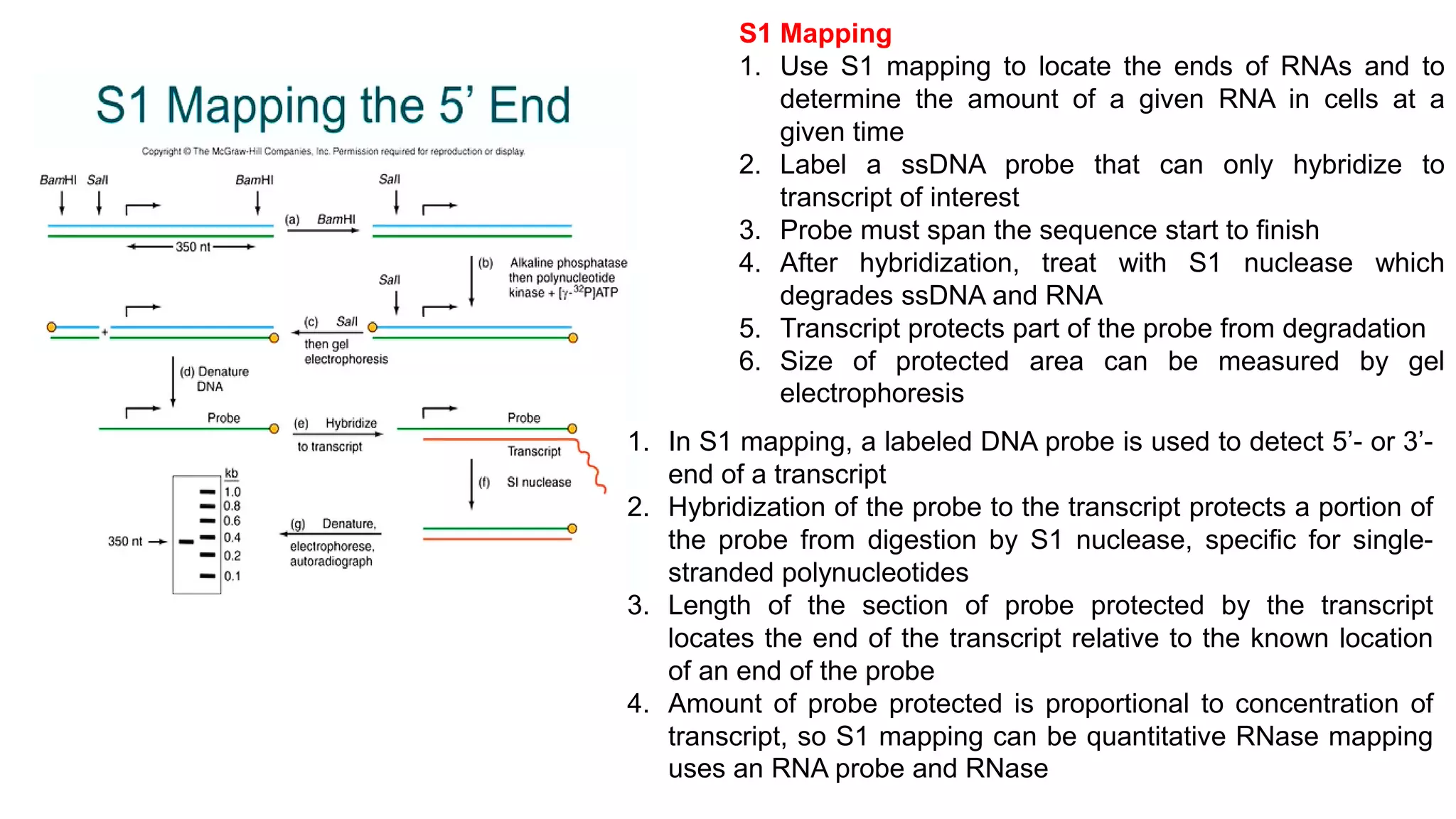
![Primer Extension
• Primer extension works to determine exactly the 5’-end
of a transcript to one-nucleotide accuracy
• Specificity of this method is due to complementarity
between primer and transcriptS1 mapping will give
similar results but limits:S1 will “nibble” ends of RNA-
DNA hybrid Also can “nibble” A-T rich regions that have
melted Might not completely digest single-stranded
regions
Primer Extension Schematic:
1. Start with in vivo transcription, harvest cellular RNA
containing desired transcript Hybridize labeled
oligonucleotide [18nt] (primer)
2. Reverse transcriptase extends the primer to the 5’-end
of transcript
3. Denature the RNA-DNA hybrid and run the mix on a
high-resolution DNA gel
4. Can estimate transcript concentration also](https://image.slidesharecdn.com/mappingandquantifyingtranscripts-220601062223-3b76982a/75/Mapping-and-quantifying-transcripts-pdf-6-2048.jpg)





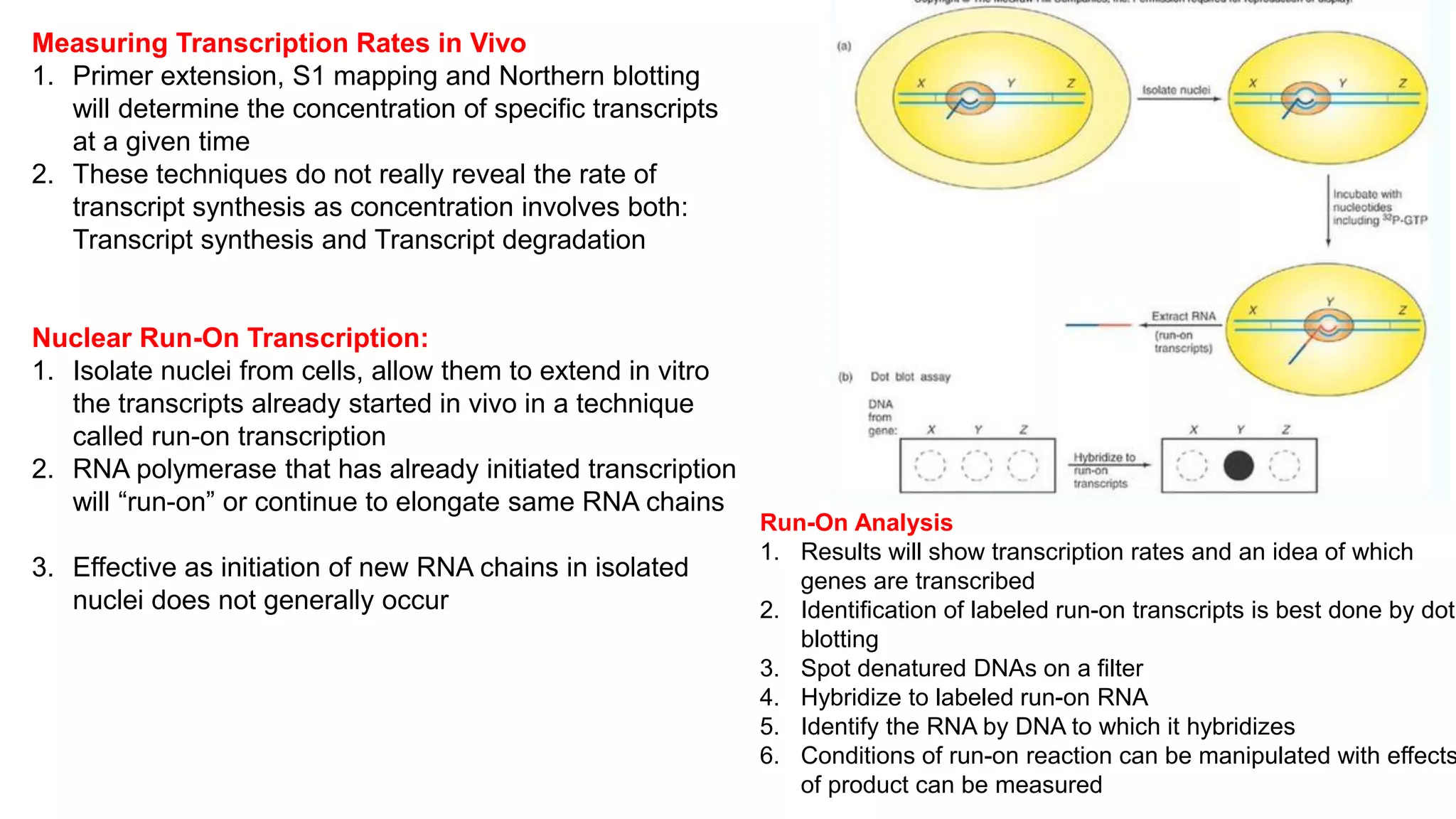


The document outlines various techniques for mapping and quantifying RNA transcripts, including northern blots, S1 mapping, primer extension, and runoff transcription. It discusses the methodologies for determining the presence, quantity, and start sites of transcripts, along with the use of g-less cassettes and nuclear run-on transcription for assessing transcription rates. Additionally, it covers methods for measuring protein accumulation such as immunoblotting and immunoprecipitation.























































































